Neuroprotectants in the Era of Reperfusion Therapy
Article information
Abstract
For decades, numerous pharmacological and non-pharmacological strategies have been evaluated without success to limit the consequences of the ischemic cascade, but more rarely the therapies were explored as add on remedies on individuals also receiving reperfusion therapies. It is plausible that these putative neuroprotectants never reached the ischemic brain in adequate concentrations. Currently, the concept of neuroprotection incorporates cerebral perfusion as an obligatory substrate upon which ischemic brain survival depends, and it is plausible that some of the compounds tested in previous neuroprotection trials might have resulted in more favorable results if reperfusion therapies had been co-administered. Nonetheless, pharmacological or mechanical thrombectomy are frequently powerless to fully reperfuse the ischemic brain despite achieving a high rate of recanalization. This review covers in some detail the importance of the microcirculation, and the barriers that may hamper flow reperfusion at the microcirculatory level. It describes the main mechanisms leading to microcirculatory thrombosis including oxidative/nitrosative stress and refers to recent efforts to ameliorate brain perfusion in combination with the co-administration of neuroprotectants mainly aimed at harnessing oxidative/nitrosative brain damage.
Introduction
A wider use of specialist stroke units, intravenous thrombolytic therapy and mechanical thrombectomy (MT) have resulted in a remarkable progress in the acute management of ischemic stroke (IS) [1-6]. However, IS still represents the first cause of permanent disability in adult people, the second single most frequent cause of death for people older than 60 years, the second most common cause of dementia, representing approximately 3% to 7% of the total health-care expenditure in high-income countries [7]. In consequence, despite the failure of numerous previous attempts during the last decades (Figure 1), there is a pressing need to continue investigating better treatments for this devastating disease [8-11].
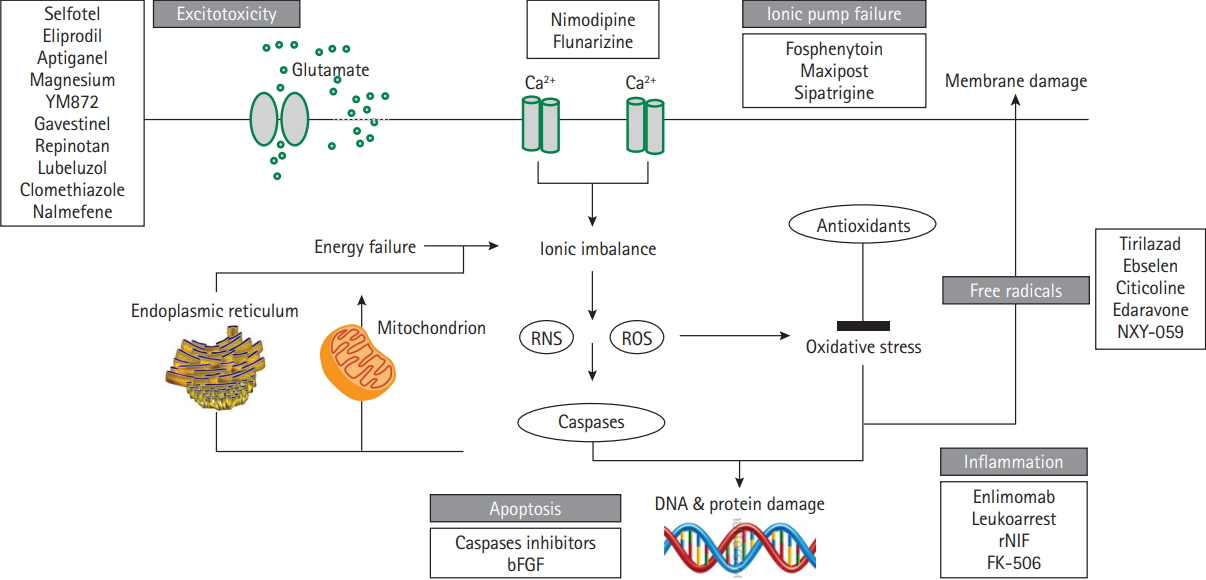
Neuroprotection in acute ischemic stroke. Main mechanisms of the ischemic cascade and some of the neuroprotectants asssessed in phase 2/3 clinical trials without beneficial effects. RNS, reactive nitrogen species; ROS, reactive oxygen species; bFGF, basic fibroblast growth factor; rNIF, recombinant neutrophil inhibitory factor; FK-506, tacrolimus.
On theoretical grounds, neuroprotection in IS is expected to be more effective the earlier the intervention is initiated but the Field Administration of Stroke Therapy–Magnesium (FAST-MAG) trial highlighted that treatment expeditiousness was not sufficient, as a treatment delay as short as 45 minutes after clinical onset failed to improve IS outcomes [12]. Therefore, other factors need to be accounted for in the rapidly changing pathophysiology of IS, including the molecular target selected for therapeutic intervention, its time of onset and persistence throughout the ischemic process, together with the pharmacokinetics and pharmacodynamics of the putative neuroprotectant [13]. As discussed below, oxidative and nitrosative stress play a major deleterious role in ischemic cell death during a large therapeutic window [14], and not surprisingly this molecular target centers the attention of a number of ongoing neuroprotectant trials. Another issue of increasing interest in stroke neuroprotection is how to maximize the rate of brain reperfusion, as it represents an obligatory substrate upon which ischemic brain survival depends [15]. Expectedly, a comprehensive approach to stroke neuroprotection would include treatment expeditiousness, a meaningful molecular target, pharmacologically fitted neuroprotectants, and strategies aimed to maximize cerebral perfusion [16,17]. This review summarizes ongoing strategies aimed at attaining a more effective neuroprotection in IS based on these principles.
Recanalization/complete reperfusion mismatch
Arterial recanalization after IS is associated with a 4- to 5-fold increase in the odds of good long-term functional outcome and a 4- to 5-fold reduction in the odds of death [18]. However, one out of four patients do not reperfuse despite complete recanalization [19-24], and one out of two develop full infarctions on regions previously hyperperfused [25], highlighting the essential role of adequate brain perfusion for the final ischemic tissue fate. In recent randomized controlled trials [1-6], less than half of patients treated with MT improved stroke outcomes regardless that three out of four had full recanalization. Arguably, the lack of clinical improvement could indicate that the recanalization occurred when a full brain infarction was established in some patients but it should be noted that only 38% of the patients obtained full reperfusion after MT, suggesting that the mismatch between complete recanalization and incomplete reperfusion explained the suboptimal efficacy of MT.
The modified thrombolysis in cerebral ischemia (mTICI) score [26] was the primary reperfusion scale used in the trials and in this scale, mTICI score 3 defines complete reperfusion of the target downstream territory (TDT) and mTICI 2b score defines restoration of more than half of the TDT, respectively (Figure 2) [27]. Current guidelines recommend achieving indistinctively mTICI 2b or 3 scores following MT, assuming that both scores maximize the probability of a good functional clinical outcome [28]. Accordingly, successful reperfusion grouped together the patients that obtained either of these two angiographic scores in the aforementioned trials. However, some studies [29-31], but not others [32,33], showed significant outcome differences between mTICI 3 and 2b scores. Recently [34], we compared the relative outcomes resulting from mTICI 2b or 3 scores at the end of MT using multivariate models adjusted for Highly Effective Reperfusion Evaluated in Multiple Endovascular Stroke (HERMES) covariates [35]. In this study, patients with mTICI 3 compared with mTICI 2b had a better mRS score at 90 days (odds ratio, 2.018; 95% confidence interval, 1.033 to 3.945), less infarct growth and smaller final infarctions, supporting a more efficient salvage of the ischemic penumbra. Therefore, mTICI 3 should always be pursued whenever technically possible, particularly in patients with salvageable tissue on brain imaging.

Main results in recent endovascular trials. Pooled percentages of the angiographic and main clinical results obtained in Randomized Trial of Revascularization with Solitaire FR Device versus Best Medical Therapy in the Treatment of Acute Stroke Due to Anterior Circulation Large Vessel Occlusion Presenting within Eight Hours of Symptom Onset (REVASCAT), Solitaire™ with the Intention for Thrombectomy as Primary Endovascular Treatment for Acute Ischemic Stroke (SWIFT PRIME), Endovascular Treatment for Small Core and Proximal Occlusion Ischemic Stroke (ESCAPE), Extending the Time for Thrombolysis in Emergency Neurological Deficits–Intra-Arterial (EXTEND-IA), and Multicenter Randomized Clinical Trial of Endovascular Treatment for Acute Ischemic Stroke in the Netherlands (MR CLEAN). mTICI, modified thrombolysis in cerebral ischemia.
Microvascular perfusion
Nearly every neuron in the human brain has its own capillary and the microcirculation represents more than 96% of the entire cerebrovascular system [36,37]. Short, middle, and long arteries branching from the pial vessels supply the human cerebral cortex by highly interconnected capillary vessels distributed in superficial, middle, and deep vascular layers [38]. Virtually all brain microvessels are constantly perfused at any time [36], but changes in the microvascular structure may hamper adequate brain reperfusion following recanalization [39]. Models of transient brain ischemia identified the lack of reflow at the microcirculation despite complete recanalization of proximal occlusions on albino rabbits [40-42]. It was attributed to spasm or swelling of the vessel wall, while increased blood viscosity was thought a secondary contributor [40]. A rich expression of thromboplastins in the brain lent support to speculate that fibrin clots formed within the capillary bed [40], while later studies also found capillary-obstructing leukocytes [43]. Clogging of the perivascular space, activation of tissue factor [44], distal microembolism from a proximal thrombus [45,46], microvascular compression by swollen astrocyte end-feet [47,48], or platelet and neutrophil adhesion, were also associated with the no-reflow. Genetic manipulation and use of anti-inflammatory agents restored effectively the no-reflow and improved stroke outcomes in experimental animals [49-53], but pharmacologic inhibition of leukocytes, platelets, or fibrin-platelet interactions were found deleterious or futile in patients with IS [54-58].
Oxidative and nitrosative stress also participates in the noreflow as the result of a rich expression of pro-oxidant nicotinamide adenine dinucleotide phosphate (NADPH) oxidases (NOX) [59] in brain endothelium, vascular smooth muscle, adventitia, and capillary pericytes [60]. In particular, pericytes are deemed to play a major role because pericytes apposed to central nervous system capillaries regulate the microcirculatory blood flow [61]. Elegant experimental studies showed that pericytes constricted at the start of ischemia because lacking adenosine triphosphate was not able to pump Ca2+ out of the cell, while suppression of oxidative-nitrosative stress relieved pericyte contraction and restored microvascular patency [62].
The efficacy of thrombolytic agents greatly depends on the quantity of intravascular thrombus [63], and therefore the no-reflow could be theoretically treated by manipulating the fibrinolytic system. Arguably, thrombi that form in the microvascular bed might be more susceptible to lyse than larger proximal thrombi. Based on this principle, the CHemical Optimization of Intraarterial Cerebral Embolectomy (CHOICE) trial is currently testing whether intraarterial alteplase is superior to placebo to improve the no-reflow at the end of MT, in a multicenter, double blind, placebo controlled phase 2 trial expected to terminate in late 2019.
Neuroprotection of the reperfused tissue: targeting the redox state
A major unifying thread in IS indicates that assorted molecular pathways converge following the onset of ischemia to produce damaging levels of free radicals and non-radicals such as peroxynitrite [64]. Sources of high concentrations of reactive species include the mitochondria [65], the activity of cyclooxygenase enzymes [66], NOX expressed by neurons [67], endothelial cells [68], and infiltrating neutrophils, and the hypoxic-dependent conversion of xanthine dehydrogenase into xanthine oxidase. Oxidative/nitrosative stress starts to rise during the early ischemic phase, followed by a much larger increment both in neurons and endothelial cells during early reperfusion [69]. For this reason, there is a growing interest in the study of putative neuroprotectants that harness the redox state in the selected population of patients treated with MT. Additionally, peroxynitrite is an especially attractive molecular target for therapeutic intervention because despite of a short half-life this compound is more toxic than other reactive species, as it crosses readily biological membranes and interacts with most critical biomolecules [70,71]. Under experimental conditions, peroxynitrite is largely generated in the ischemic penumbra where it builds up for about 6 to 12 hours [72]. Once formed, peroxynitrite facilitates the demise of the penumbra by lipid peroxidation, mitochondrial damage, protein nitration and oxidation, depletion of antioxidant reserves, activation or inhibition of various signaling pathways, DNA damage, and blood-brain barrier (BBB) breakdown [73].
Clinical trials
Uric acid (UA) is a potent peroxynitrite scavenger [74,75], and the evaluation of its putative neuroprotectant effects is at an advanced phase of clinical development [ UA is the end product of the catabolism of purines, the most abundant natural antioxidant in humans and several-fold more effective than previous antioxidant compounds tested in IS [76]. Following brain ischemia there is a rapid reduction of the endogenous levels of UA that reach the nadir within 6 hours after clinical onset [77]. In a rat thromboembolic model, UA conveys synergistic neuroprotection with alteplase [78], and in rodents with transient or permanent ischemia UA decreases the production of reactive oxygen species (ROS), reduces infarct volume and improves outcome [78-81]. UA therapy also prevents the production of superoxide, nitric oxide, nitric oxide synthase (NOS), endothelial NOS, neuronal NOS, and interleukin-18 in the ischemic arterial wall, and protein nitrosylation in the brain despite a poor BBB permeability [81], suggesting UA mediated neuroprotection mainly derives from the cerebral vasculature. Recently, the Efficacy Study of Combined Treatment With Uric Acid and r-tPA in Acute Ischemic Stroke (URICO-ICTUS) trial confirmed in 421 patients with IS the safety of UA therapy in combination with alteplase, and a significant reduction in the risk of early ischemic worsening (Figure 3) [82]. Whilst the trial was unable to demonstrate a significant benefit in the whole study population, it significantly achieved the primary outcome of the trial in predefined subgroup analyses, including in women [83], patients of either sex having stress hyperglycemia [84], and patients that received MT as part of the initial reperfusion therapy [85]. In the latter group, UA therapy showed a 19% absolute increase in the proportion of good outcome compared with placebo despite similar rates of complete reperfusion at the end of MT. The Uric acid in Reperfusion Injury Control (URIC) trial has been recently planned to validate the URICO-ICTUS results in a larger population of patients treated with MT.
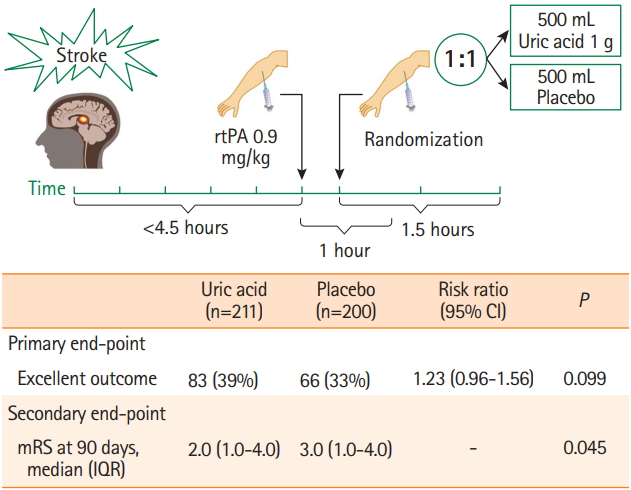
Study flow and main outcomes in Efficacy Study of Combined Treatment With Uric Acid and r-tPA in Acute Ischemic Stroke (URICO-ICTUS) trial 3. All the patients received alteplase within 4.5 hours after symptoms onset and were randomized 1:1 to a 90 minutes intravenous infusion of uric acid or placebo. The primary outcome—rate of patients with a modified Rankin Scale (mRS) score 0 to 1, or 2 if this was the premorbid functional status—showed a non-significant statistical trend. Main secondary outcomes, including the median mRS (P=0.05) and the mRS shift (odds ratio, 1.40; 95% confidence interval [CI], 0.99 to 1.98; P=0.05) were statistically significant. rtPA, recombinant tissue plasminogen activator; IQR, interquartile range.
The ongoing Safety and Optimal Neuroprotection of neu2000 in Ischemic Stroke With Endovascular reCanalization (SONIC) trial (NCT02831088) aims to provide proof-of-concept for use of Neu2000 as an adjunct neuroprotective agent together with state-of-the art endovascular therapy (EVT) in patients with IS. Neu2000 (a derivative of sulfasalazine) is a novel, multitarget neuroprotectant that combines modest, subtype-selective (NR2B) blockade of N-methyl-D-aspartate (NMDA) receptors with potent scavenging of ROS [86,87]. It has been suggested that the prior failure of more potent, and subtype unselective NMDA antagonist drugs in stroke trials may be partly due to excessive reduction of intracellular free calcium levels and consequent enhancement of ischemic neuronal apoptosis [88]. Neu2000 seeks to blunt acute excitotoxicity without risking this downside, and then to additionally target downstream free-radical damage, prominently triggered by vascular reperfusion. The neuroprotective potential of Neu2000 has been demonstrated in preclinical animal stroke models with a favorable efficacy and therapeutic window (administration in delayed time window) profile [88,89].
Plasma glucose is another important stressor in experimental models of focal cerebral ischemia/reperfusion which may contribute to the worsening the fate of stroke [90,91]. Unlike originally incriminated [92], extracellular lactate accumulation is not a crucial determinant of brain injury in experimental hyperglycemia [93], for prevention of tissue acidosis does not avoid brain tissue damage under hyperglycemic conditions [94]. Rather, oxidative stress and inflammatory mechanisms are increasingly involved in hyperglycemia-induced endothelial injury in transient ischemia [95], while antioxidants have shown experimentally to attenuate this damage [96]. Inactivation of the glucose-dependent nicotinamide adenine dinucleotide phosphate oxidase enzyme blocks neuronal ROS production and negates the deleterious effects of hyperglycemia [97]. As discussed above, in URICO-ICTUS, UA therapy was more effective in patients with stress hyperglycemia than in normoglycemic patients at stroke onset, suggesting that a greater availability of glucose increased the formation of free radicals in the ischemic penumbra [98]. Currently, the Intensive Insulin Therapy With Tight Glycemic Control to Improve Outcomes After Endovascular Therapy for Acute Ischemic Stroke trial (NCT02054429) is assessing the safety and efficacy of lowering glucose (blood sugar), in addition to MT, to improve 90-day functional and neurological outcomes in comparison to standard glycemic care in 100 patients with IS. The study will involve treatment of 100 (50 intensive insulin therapy and 50 standard glycemic control) non-diabetic patients presenting within 8 hours of acute IS who have undergone EVT.
Preclinical studies
Other compounds with redox based properties have shown neuroprotective effects in experimental models of brain ischemia but have not reached yet a phase of clinical testing at the bedside. Amongst these compounds, 3, 4-dihydro-6-hydroxy-7-methoxy-2,2-dimethyl-1(2H)-benzopyran (CR-6) is a synthetic, structurally simpler, analogue of vitamin E, with the additional capacity to scavenge nitrogen-reactive species, that has shown neuroprotective effects in brain ischemic rats [99]. Interestingly, CR-6 was only effective in animals that developed hyperemia at reperfusion, highlighting the relevance of reperfusion for redox based neuroprotection [99].
Another treatment option supports directly targeting the right enzymatic source of ROS rather than applying non-specific antioxidants after radicals have already been generated [100]. It has also been proposed a more targeted supply of antioxidants to key subcellular locations, for example the mitochondria [101]. A most attractive candidate is the NOX family, the only known enzymes solely dedicated to ROS production. NOX are multicomponent protein complexes containing a catalytic subunit that transfers electrons from NADPH to oxygen, thereby forming ROS. There are seven isoforms with different tissue distribution and whose components are separated between the cytosol and the plasma membrane [102]. Under basal conditions, these components are kept apart rendering the enzyme inactive, but becoming activated at the onset of reperfusion [59]. NOX4 is the most widely distributed NOX isoform in the vasculature and its expression is higher in cerebral arteries compared with peripheral blood vessels [103]. After an ischemic insult, NOX4 is upregulated in neuronal cells and brain microvascular endothelial cells [104], while NOX2 is synthesized by microglial cells and recruited phagocytes [105]. Controversies remain whether NOX2 [106], or NOX4 [107] plays the most relevant role in stroke pathophysiology, although deletion of both NOX2 [108], and NOX4 [109], have been associated with reduction in infarct size, while overexpression of NOX4 was associated with larger infarcts [110].
Compounds with a low specificity to inhibit NOX have also been investigated in experimental models of cerebral ischemia/reperfusion injury, including diphenylene iodonium [111], the serine protease inhibitor 4-(2-aminoethyl) benzenesulfonyl fluoride [112], and 3-hydroxy-3-methyl-glutaryl reductase inhibitors (statins) [113]. L-arginine is a NOS substrate and precursor to nitric oxide, as well as an enhancer of cerebral blood flow [114]. In 10 experimental studies of the effects of L-arginine on behavior and/or infarct volume the results were heterogeneous and inconclusive [115,116].
Another approach is to boost the activity or preventing the inhibition of intrinsic antioxidant defenses, including the recently discovered master regulators of antioxidant defenses nuclear factor-erythroid 2-related factor 2 (Nrf2) or peroxisome proliferator-activated receptor-γ coactivator 1α [117]. Recently, Nrf2 has shown cytoprotective mechanisms in the retina in response to ischemia-reperfusion injury and pharmacologic induction of Nrf2 has been posited as a new therapeutic strategy for retinal ischemia-reperfusion [117].
The toxicity of peroxynitrite may also be prevented indirectly by strategies that block nitric oxide or superoxide generation, or by using direct approaches including peroxynitrite or peroxynitrite-derived radical scavengers, and peroxynitrite decomposition catalysts [118]. In addition to the effects of UA discussed previously, other indirect inhibitors of peroxynitrite include N-tertbutyL-α-phenylnitrone (PBN), the NOS inhibitor Nω-nitro-L-arginine (L-NA), and the BBB-impermeable NOS inhibitor L-N5-(1-iminoethyl)-ornithine (L-NIO). These three agents have shown to prevent pericyte constriction, restore the patency of capillaries, and improve tissue recovery in brain ischemic mice [62,119]. Interestingly, a similar degree of neuroprotection and inhibition of 3-nitrotyrosine formation was found with the BBB-impermeable L-NIO, as compared to BBB-permeable L-NA and PBN [62], emphasizing that the vasculature is a major source of radicals during reperfusion and suggesting that vascular-bed strategies can result in an effective neuroprotection.
Peroxiredoxins (Prxs) belong to a ubiquitous family of peroxidases that have been shown to readily and catalytically reduce peroxynitrite to nitrite through the reaction of the peroxidatic cysteine. There is emerging evidence that the antioxidant Prxs become inactivated during an ischemic episode [120], and Prxs have also been shown to be protective in several models of ischemia in vitro and in vivo through overexpression studies [121].
Synthetic molecules that react directly and catalytically decompose peroxynitrite include metalloporphyrins of iron and manganese. Peroxynitrite decomposition catalysts such as 5,10,15,20-tetrakis(N-methyl-4′-pyridyl) porphyrinato iron III (5+) (FeTMPyP) and 5,10,15,20-tetrakis(4′-sulphonatophenyl) porphyrinato iron III (3–) (FeTPPS) can reduce infarction size, brain edema, and neurological deficits when administered at 6-hour after experimental ischemia [122], and improve vascular dysfunction in mild hyperglycemic ischemic rats [123]. The catalyzed reaction is a net isomerization of peroxynitrite to nitrate. The efficacy of some of the manganese-based compounds, primarily MnTE-2-PyP (5+), have been tested in vivo and have shown to exert marked neuroprotective effects against focal ischemic insults [124]. The fact that the compound remained effective even when it was given up to 6 hours after ischemia implicates the role of peroxynitrite in the delayed ischemic death processes. A related compound, Mn (III) tetrakis (N-N′-diethylimidazolium-2-yl) porphyrin (AEOL-10150) has demonstrated efficacy in preclinical models of stroke in rats and mice although the greater effects were observed following intracerebroventricular administration, although more modest effects were seen after intravenous injection, which may limit its clinical use [124].
Conclusions
For many years the field of neuroprotection in IS has yielded a long record of frustrating clinical results but also great advances in our understanding of the mechanisms unleashed during the ischemic cascade including the major role of oxidative and nitrosative stress within the brain parenchyma and in the microcirculation. Most previous neuroprotectant trials gave limited emphasis to the importance of full reperfusion to salvage the ischemic penumbra, casting doubts at what concentration the putative neuroprotectant did reach the ischemic neurovascular unit. The advent of MT offers an excellent opportunity to reverse this situation and opens a new scenario where combined strategies might maximize the arrival of the neuroprotectants to the tissue at risk and also the reperfusion of the microcirculation. Hopefully, the new strategies which are currently undergoing might contribute to declare soon that the inability to effectively protect the ischemic brain at the bed side is a hardship of the past.
Notes
Disclosure
Ángel Chamorro is inventor of the patent “Pharmaceutical composition for neuroprotective treatment in patients with ictus comprising citicoline and uric acid.”
Acknowledgements
Supported by FIS PI15/00430, co-financed by the ISCIII-Subdirección General de Evaluación and Fondo Europeo de Desarrollo Regional (FEDER) and “CERCA Programme/Generalitat de Catalunya.”