Introduction
Alcohol is consumed widely throughout the world. In Western countries, abstainers constitute the minority while most have a light to moderate alcohol intake (drinking a max of 2 and 3 drinks per day for women and men). Harms associated with this habit are therefore of huge public health and of potential clinical concern.
The association between alcohol intake and stroke risk has been addressed in a number of epidemiological studies, with differing results; especially controversy exists whether drinking in moderation confers a reduced risk of ischemic stroke, in accordance with some studies [1-5], and increased risk of haemorrhagic stroke [1,6]. Moderate alcohol intake was not associated with stroke, ischemic or haemorrhagic, in most studies [3,7-11] which however in a recent meta-analysis was summed up to a significantly reduced pooled relative risk (RR) of ischemic stroke for a low intake (<15 g equalling approximately 1.3 drinks per day) compared with abstaining [12]. However, especially the largest individual studies that took part of the meta-analysis contributed with insignificant findings with statistical estimates near 1.0, while studies of smaller size had results further away from the null, all in all indicating that a “protective effect” of alcohol on ischemic stroke is either small or that bias is present. Another meta-analysis also concluded that light to moderate drinking (≤2 drinks/per day) was associated with a reduced risk of ischemic stroke and heavy drinking (>4 drinks/per day) was associated with increased risk of both ischemic and haemorrhagic stroke [13].
Generally, when studying health effects associated with drinking in moderation, one has to rely on the participants’ own report, implying possibility of reverse causality and confounding. Disentangling causal effects of alcohol from effects of related life style factors is likely to be especially difficult at the low to moderate intake level, as difference in true risk is small compared with the reference group of non-drinkers. Complicating such comparison further is the facts that non-drinkers often constitute a heterogeneous group of individuals who abstain by simple preference and individuals who abstain due to health or socioeconomic reasons. For instance, relatively more abstainers than light drinkers have chronical diseases and low socioeconomic position [14,15].
An attempt to address the problems associated with self-reported information is to use the Mendelian randomization design, a method that uses genetic variants as instrumental variables for the exposure, instead of the exposure itself [16,17]. For alcohol, functional variants in genes encoding alcohol dehydrogenase (ADH), responsible for the majority of the body’s alcohol degradation, are associated with extent of alcohol intake: individuals who have the fast alcohol degrading alleles ADH1B*2 and ADH1C*1 consume less alcohol than those with slow degrading alleles ADH1B*1 and ADH1C*2, which persists across age groups from early adulthood to old age, and observed in various populations [18-22]. In general, instruments valid for Mendelian randomisation (MR) analysis should be associated with the exposure of interest (in this case, amount of alcohol intake) and should influence the outcome through that exposure only [23]; i.e., there should be no pleiotropic effects. For the alcohol dehydrogenase 1B and 1C (ADH1B and ADH1C) genotypes, no pleiotropic effects have been identified [24]. All taken together, ADH genotypes are valid candidates for unbiased instruments of lifelong alcohol consumption.
In this study we tested the hypothesis that alcohol intake is associated observationally and causally through genetic variation with risk of any, ischemic and haemorrhagic stroke. For this purpose, we used data from two large population-based cohorts with information on self-reported alcohol consumption and genetic variations in ADH1B and ADH1C as instrumental variables.
Methods
Study populations
The Copenhagen General Population Study (CGPS) is an ongoing prospective study of the Danish general population initiated in 2003. To be eligible, participants had to be of Danish descent (i.e., the participant and both parents were born in Denmark and were Danish citizens). Among individuals living in counties of greater Copenhagen aged 20 to 40 years, 25% were invited, whereas all eligible people aged >40 years were invited. The response rate was 42%. When the present study was initiated, 84,720 individuals had been included.
The Copenhagen City Heart Study (CCHS) was initiated in 1976 where a random sample of the Danish general population above age 20 years living in the Center of Copenhagen was invited to participate (number of participants, 14,223; response rate, 74%). This examination was followed by three examinations; a second examination in 1981 to 1983, where all previously invited plus 500 new individuals aged 20 to 24 years were invited (n=12,698, 70%); a third examination in 1991 to 1994 where all previously invited plus 3,000 new individuals aged 20 to 49 years were invited (n=10,135, 61%); and a fourth examination in 2001 to 2003 where all previously invited plus an additional sample of 1,040 individuals aged 20 to 29 years were invited (n=6,238, 50%). Eligibility criterion was Danish citizenship, and the participants does not reflect the ethnic admixture of Copenhagen (for instance, the proportion of inhabitants with foreign citizenship was 8% in 1994). However, even a few participants of foreign ethnicity could confound our results since the ADH1B*2 allele has shown considerable population stratification [25]. Hence, information on birth place as recorded in the Danish Civil Registration System [26] was obtained, and participants who were born in Asia, Africa, the Middle East, South America, and Greenland were excluded (n=243).
We used data from CCHS 1991 to 1994 and 2001 to 2003 examinations (n=11,365) and the CGPS 2003 to 2012 examination (n=84,720) [27-29]. Data were collected following similar principles. Participants filled in a questionnaire that was reviewed by an investigator on the day of attendance, had a physical examination performed, and had blood samples drawn for biochemical measurements and DNA extraction. Written informed consent was obtained. The Danish Ethics Committee approved the studies (No. 100.2039/91 and H-01-144/01).
Assessment of alcohol consumption
Participants reported their consumption of beer (in bottles), wine (in glasses), and spirits (in units) with response categories of “never/ hardly ever,” “monthly,” “weekly,” or “daily” and number of drinks per week. The total number of drinks per week was calculated by summing up the individual types of alcohol and was categorised as in previous studies [30]: 1 to 6, 7 to 13, 14 to 20, 21 to 27, 28 to 34, and ≥35 drinks per week with 1 drink equalling 12 g alcohol. For women, the highest category was ≥28 drinks per week.
Previously, we have shown that stepwise increments of the self-reported alcohol consumption consistently corresponded to stepwise increments in factors expected to correlate with amount of alcohol consumption (alanine aminotransferase, γ-glutamyl transpeptidase, erythrocyte volume, and alkaline phosphatase), speaking in favour of the validity of self-reported alcohol consumption [18].
ADH1B and ADH1C genotypes
DNA was isolated from full blood and stored at -45℃. Participants were genotyped for the ADH1B genotype (rs1229984; Arg47His) and ADH1C genotype (rs698; Ile349Val) by Nanogen Technology (Carson City, NV, USA) [31,32] and TaqMan assays (Applied Biosystems, Foster City, CA, USA) [19]. Genotypes were in Hardy-Weinberg equilibrium in both studies (P>0.05). Genotypes ADH1B* 2/1 and ADH1B*2/2 were grouped because of few participants with these genotype. ADH1B and ADH1C genotypes were combined and ranked according to expected enzyme activity, ranging from most to least active (1, ADH1B*2/x+ADH1C*1/1; 2, ADH1B*1/1+ADH1C*1/1; 3, ADH1B*1/1+ADH1C*1/2; 4, ADH1B*1/1+ADH1C*2/2). Additionally, we used regression analysis with alcohol intake as a dependent variable and sex and genotypes as independent variables to test how much of the variance in alcohol consumption was explained by the ADH1B and ADH1C genotypes.
Assessment of ischemic and haemorrhagic stroke
Information on diagnosis of stroke was obtained from the Danish National Patient Register that holds records of all Danish hospital admissions since 1978 and further outpatient and emergency ward contacts since 1995, and from the Danish Causes of Death Registry which contains information on causes of death for all deceased in Denmark [33-35]. In both registers, records are classified according to World Health Organization (WHO) International Classification of Diseases (ICD). Records with the following codes implying stroke were extracted: ICD-8 431 to 438 and ICD-10 I60 to I68 and G45.
All diagnoses of stroke were validated individually. Records were requested from general practitioners and hospitals and validated by two experienced medical doctors independently, blinded to the test results [36,37]. Ischemic stroke was defined according to WHO criteria as rapidly developing signs of focal or global disturbance of cerebral function lasting >24 hours (unless interrupted by death), with no apparent nonvascular cause [38]. Haemorrhagic stroke and subarachnoidal hemorrhage were excluded from the ischemic stroke group. To distinguish among infarction, intracerebral hemorrhage, and subarachnoidal hemorrhage, either computed tomography or magnetic resonance imaging scan, spinal fluid examination, autopsy, or surgical description were required. If the scan did not show infarction or hemorrhage, but symptoms in accordance with stroke criteria, that event was diagnosed as ischemic stroke. The diagnosis of ischemic and haemorrhagic stroke was not applied in cases in which a scan revealed signs of prior cerebrovascular disease, but without history of any symptoms, if symptoms were nonfocal, or if symptoms lasted <24 hours [39]. Alcoholic liver cirrhosis was ascertained similarly from the Danish registries using ICD-8 code 571.0 and ICD-10 code k70.3.
Statistical analysis
Of the initial sample of 89,675 individuals, we excluded those with stroke prior to baseline (n=2,737), missing information on alcohol consumption (n=4,499) and covariates (smoking [n=2,573]), school education (n=247), body mass index (BMI) (n=511), and leisure time physical activity (n=562). The final analytic sample therefore consisted of 78,546 men and women (where genotype information was available for 74,632 individuals) (Table 1).
Variables considered being potential confounders included the following: age; school education (<8, 8 to 11, and >11 years of education, corresponding to lower primary school, higher primary school, and secondary school); leisure time physical activity (sedentary, light, moderate, and vigorous); smoking (never smoker, former smoker, and current smoker of 1 to 14, 15 to 24, and >24 g of tobacco/day); BMI (linearly); family history of cardiovascular disease (either biological parent having had myocardial infarction or stroke); diabetes (by self-report), angina pectoris (by self-report), hypertension (by self-report); ischemic heart disease (hospital diagnosis before baseline, yes/no); congestive heart failure (hospital diagnosis before baseline, yes/no); angina pectoris (by self-report); heart medication (by self-report, yes/no); and use of cholesterollowering medication (by self-report, yes/no). Analyses were conducted using Stata version 12.1 (StataCorp, College Station, TX, USA).
P-values for trend between genotypes and baseline characteristics were calculated using chi-square test for categorical variables and Kruskal-Wallis test for continuous variables. For this purpose, genotypes were assigned with values reflecting the effect of genotypes on alcohol consumption.
Participants accrued person-time from baseline until time of stroke (n=2,335), death (n=7,524), emigration (n=468), or end of follow-up (n=68,161), whichever occurred first. Data from the CGPS and CCHS were combined after having ensured that results were similar in the two studies separately. This was tested formally in a nested log-likelihood test (P=0.16 for any stroke). Power calculation for the MR analysis were conducted using Stata procedure power logrank. Analyses were conducted following the four steps below, in accordance with the Mendelian randomization study principle [17,40].
Self-reported alcohol intake and stroke
We tested whether alcohol intake was associated with stroke using Cox proportional hazards models with delayed entry. Age (in days) was used as the underlying time axis to ensure maximal adjustment for confounding by age. The Cox proportional hazards assumption was examined graphically by plots of log (time) by log (-log [survival probability]) and statistically by introducing interaction terms between age and alcohol intake in the model. No violations were detected (P>0.05).
Association between ADH1B and ADH1C genotypes and alcohol intake
Second, we tested whether ADH1B and ADH1C genotypes and their combinations were associated with alcohol intake. This was done in linear regression analysis, using the Cuzick’s extension of the Wilcoxon rank sum test to calculate P-value for trend.
Alcohol and stroke: using ADH1B and ADH1C as instruments for alcohol intake
Third, we tested whether genotype combinations were associated with risk of stroke using Cox regression, modelled as described in 'step 1.'
Alcohol and stroke: observational versus genetic estimates
Fourth, to test a potential causal association between alcohol and stroke, we performed instrumental variable analysis with a two-stage regression model using genotype combinations as instruments to estimate the effect of per additional genetic drink per day on risk of stroke [16]. The first stage was a linear regression of genotype combinations on alcohol consumption. F-statistics >10 indicates sufficient statistical strength to carry out statistically valid instrumental variable analysis, which was confirmed (F=22.0) [41]. The second stage was a logistic regression of alcohol intake determined by genotype combinations (generated in the first stage) on stroke to calculate genetic risk ratios. For comparison, we calculated hazard ratios (HRs) for the association per additional observational drink per day and risk of stroke.
Results
Baseline characteristics of the 37,344 men and 44,935 women are listed in Table 1, in total and by categories of alcohol intake. Further, P-values are shown for associations between each characteristic and alcohol consumption, and between characteristics and ADH1B and ADH1C genotype combinations. Most characteristics were associated with alcohol consumption. In contrast, none of the baseline characteristics were associated with genotypes after Bonferroni correction for 22 comparisons. This illustrates likely substantial confounding in observational analyses, but none detected in causal, genetic analyses. The mean follow-up duration was 7.9 years. During the study period, 2,535 incident cases of ischemic and haemorrhagic stroke occurred.
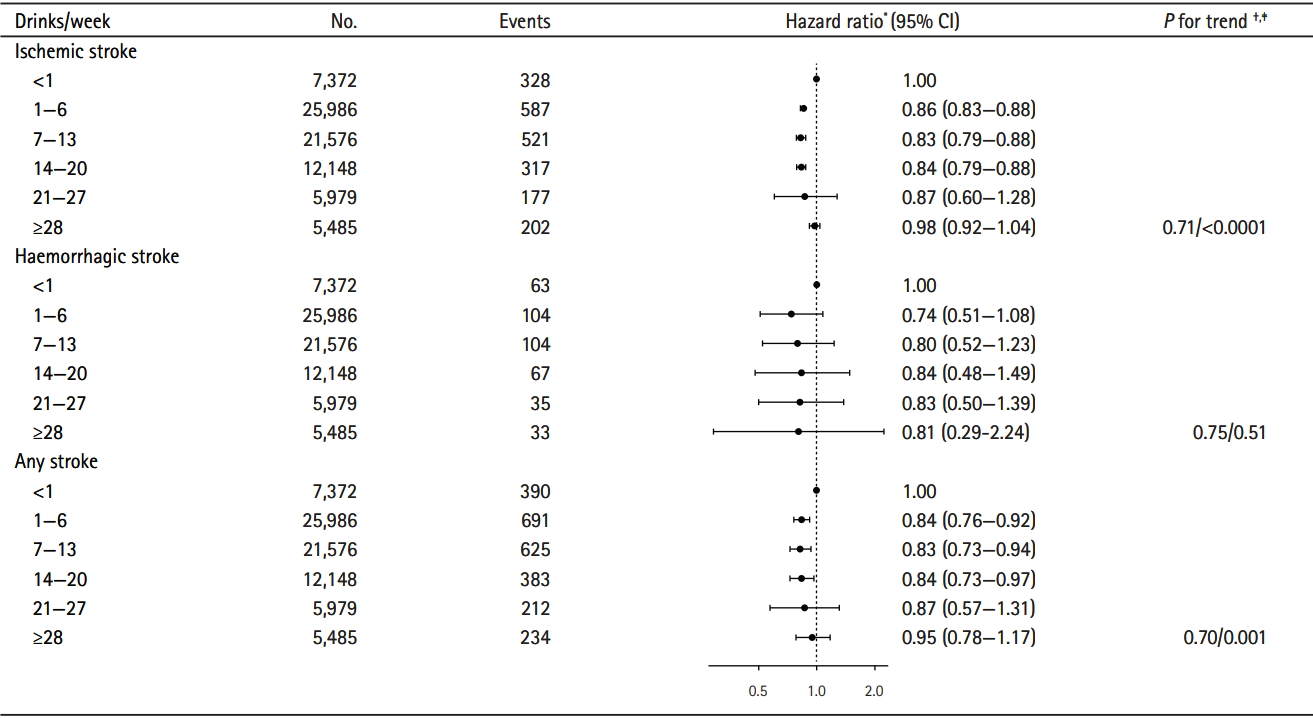
Men and women who consumed 1 to 20 drinks/week had reduced risk of any stroke (ischemic and haemorrhagic stroke combined) compared to men and women consuming less than 1 drink/week (Figure 1). No significant association was seen among individuals consuming 21 drinks/week or more. The HRs for any stroke associated with drinking 1 to 6, 7 to 13, and 14 to 20 drinks/week versus less than 1 drink/week were 0.84 (95% confidence interval [CI], 0.76 to 0.92), 0.83 (95% CI, 0.73 to 0.94), and 0.84 (95% CI, 0.73 to 0.97), respectively, and the P-value for linear trend was <0.0001. The pattern was similar for ischemic and haemorrhagic stroke, but only statistical significant for ischemic stroke.
Evidence of a nonlinear relationship between alcohol intake and total stroke was further supported by Figure 2 showing HRs for any stroke by increasing alcohol intake with <1 drink/week as reference. Indeed, alcohol intake of 1 to 20 drinks/week was associated with reduced risk of total stroke. Heavy alcohol intake tends to be associated with an increased risk of any stroke, indicating a j-shaped association (Figure 2); however, the association between high alcohol intake and risk of any stroke was not statistically significant (Figure 1).
Further, testing for non-linear trend using the mean alcohol intake for each genotype combination (Table 2), and modelling this amount with a linear and a square term analogous to test for trend as presented in Figure 1 was statically insignificant (P=0.40 and P=0.42 for linear and squared terms). Men and women with the ADH1B and ADH1C slow and intermediate genotypes had higher alcohol consumption than those with the ADH1B and ADH1C fast genotype (Table 2). For example, individuals with the ADH1B and ADH1C slow and intermediate genotypes on average drank 22% and 25% more alcohol than individuals with the ADH1B and ADH1C fast genotype. Result from the regression analysis showed that 10% of the variation in alcohol consumption was explained by the ADH1B and ADH1C genotypes.
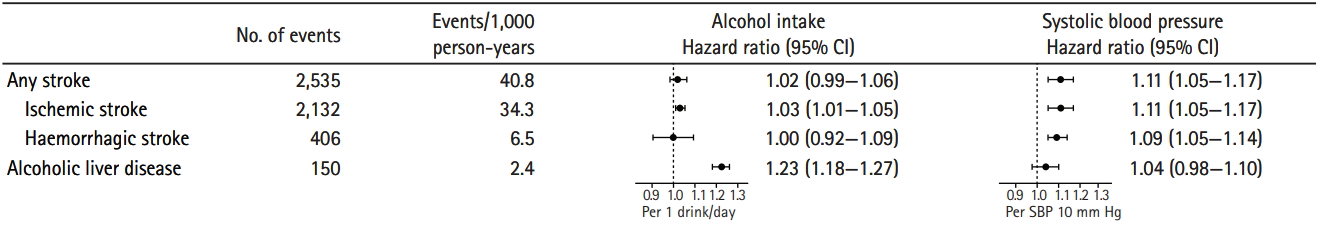
The risk of total stroke was similar among individuals with genotypes coding for high compared with low alcohol consumption: using the ADH1B and ADH1C genotype combination associated with the lowest alcohol consumption (on average 8.9 drinks/week) as reference, the HR for any stroke for the genotype combination that was associated with the highest alcohol consumption (11.1 drinks/week) was 1.15 (95% CI, 0.66 to 2.02) (Table 2). Results obtained in the present study were in keeping with results from the literature, when examining already established association. Hence, increasing alcohol consumption (per 1 drink/day) was positively associated with risk of alcoholic liver disease (HR, 1.23; 95% CI, 1.18 to 1.27), but not associated with risk of any stroke (HR, 1.04; 95% CI, 0.98 to 1.20), and increasing blood pressure (per systolic 10 mm Hg) was not associated with risk of alcoholic liver disease (HR, 1.04; 95% CI, 0.98 to 1.10) but positively associated with risk of any stroke (HR, 1.11; 95% CI, 1.05 to 1.17) (Figure 3).
Discussion
In this prospective cohort study, observational (self-reported) alcohol consumption was associated with lower risk of any stroke (ischemic and haemorrhagic stroke combined) in men and women consuming 1 to 20 drinks/weeks. Heavy alcohol intake if anything tended to be associated with an increase in the risk of any stroke, indicating a j-shaped association. However, no significant association was seen among individuals consuming 21 drinks/week or more. A similar pattern was seen for ischemic and haemorrhagic stroke separately, but this was only significant for ischemic stroke with the most accumulated endpoints. From analyses using genetic variation in alcohol metabolizing genes (ADH1B and ADH1C) as instrumental variables for alcohol consumption, we found no evidence to support causality of the observational findings. When examining already established associations for the included endpoints, the results obtained in the present study are in agreement with results from the literature.
Our results are in accordance with the results in a recent meta-analysis of 20 prospective cohort studies showing that low alcohol intake (<15 g/day, corresponding to less than 1 standard drink), compared with no alcohol intake, was associated with a reduced risk of any stroke (RR, 0.85; 95% CI, 0.75 to 0.95). Further, compared with no alcohol intake, moderate alcohol intake (15 to 30 g/day, corresponding to approximately 1 to 2 standard drink) showed no significant association with total stroke (RR, 1.01; 95% CI, 0.93 to 1.09) whereas heavy alcohol intake was associated with an increased risk of any stroke [12]. Low alcohol intake was likewise associated with a reduced risk of ischemic stroke. However, no association between moderate and heavy alcohol consumption and ischemic stroke was evident. Last, the meta-analysis found no association between alcohol intake at any level and risk of haemorrhagic stroke. Another meta-analysis of 27 prospective cohort studies found that light to moderate alcohol consumption (≤2 drinks/day) was associated with a reduced risk of ischemic stroke but not associated with haemorrhagic stroke subtypes [13]. Further, heavy drinking (>4 drinks/day) was found to be associated with all stroke types.
Plausible explanations
Our result and the dose-response meta-analysis suggest that a potential nonlinear relationship is present between alcohol intake and risk of any stroke, and that an alcohol intake of 1 to 20 drinks/week is associated with a reduced risk of any stroke. Hence, the association seems to be j-shaped. The divergent dose-response relationships between alcohol consumption and risk of ischemic and haemorrhagic stroke subtypes suggest that different mechanisms underlie associations with the different stroke types. Alternatively, the slightly different results may be due to fewer haemorrhagic stroke events compared with ischemic stroke events, and thus the statistical power could be too low for reliable risk estimates for haemorrhagic stroke alone. Heavy alcohol intake may be associated with an increased risk of haemorrhagic events because heavy alcohol intake potentially results in high blood pressure, reduced platelet aggregation, and enhanced fibrinolysis [12,13]. Further, alcohol consumption is associated with increased high density lipoprotein cholesterol levels, decrease platelet aggregation, increase fibrinolysis, and decrease plasma fibrinogen levels and this might help explain the lower risk of ischemic stroke [12,13].
Strengths and limitations
From analysis using ADH genotypes as instrument for alcohol consumption, we did not obtain results that supported causality of the observational finding. The strength of using genetic instruments for lifestyle exposures is that confounding from associated risk factors and reverse causation are unlikely, and causal interpretation of results may be more straight forward than causal interpretation of observational findings [17]. Further, selection bias due to non-participation is less likely to affect results for genetic instruments [42] whereas as for results obtained for self-reported alcohol intake, bias cannot be ruled out: if alcohol intake was associated with non-participation and with risk of stroke, results would be influenced [43]. Hence, heavy drinkers who participate may be at better health than heavy drinkers in the underlying population, whereas light and moderate drinkers who volunteer may be more representative of moderate and non-drinkers drinkers generally. Ultimately, such a mechanism would lead to more accurate results among the light to moderate drinkers and underestimated results among the heavy drinkers.
A limitation is that the ADH1B and ADH1C genotypes are not strong instruments, meaning that the influence on the amount of alcohol consumption is limited; 10% of the variation in alcohol intake was explained by the ADH1B and ADH1C genotypes in this population. Hence, the lack of a positive finding does not preclude that alcohol is causally associated with stroke. Further, using these genotypes as instruments implies a linear model between genetic alcohol consumption and the risk of stroke; if the association between alcohol and risk of stroke for instance is indeed threshold-shaped, then the genetic instrument has little power to pick it up.
Further, another limitation is lack of power in the MR analysis. We computed the lowest HR that can be picked up with the present set-up, for the ADH1B/ADH1C combination with the highest alcohol intake (i.e., the 1/1;1/2 group, please refer to Table 2), with a 90% power and a significance level of 0.05: this HR is 0.65, meaning that we do not have the adequate power to assess a HR of 0.84.
The reason that ADH genotypes is associated with alcohol consumption is most likely that—for a given level of intake—individuals with fast alcohol degradation have higher levels of acetaldehyde leading to unpleasant symptoms such as nausea and flushing compared with individuals with slow degradation, causing them to drink less. Thus, peak levels of acetaldehyde, known to be carcinogenic and playing important roles in the development of alcohol-related diseases [44], will be higher in fast compared to slow metabolisers, meaning that by using ADH1B and ADH1C genotypes as instruments the difference in amount of alcohol cannot be studied independently of difference in acetaldehyde levels, which constitute a final limitation of this approach.
A candidate for another instrument of alcohol intake to be used in future MR studies is the recently identified single nucleotide polymorphism (SNP) in Klothoβ (KLB) gene (encoding β-Klotho), demonstrated to have a role in the regulation of alcohol drinking, observed in independent samples [45]. While the mechanism linking the KLB gene with alcohol intake is still unknown, making the potential for using it as an instrument for alcohol intake unsure it is a promising candidate to be used alone or in combination with ADH1B and ADH1C genotypes forming stronger instruments. Last, it was not possible to separate former drinkers from abstainers and hence, some drinker misclassification might have occurred.
Study strengths include the large number of individuals and stroke endpoints, and that the hospital register information on the stroke diagnosis was highly sensitive and specific [36,37]. Further, the National Danish Patient Register covers all hospitalizations in Denmark including outpatients and emergency wards since 1995, and only if participants were treated in another country or by a general practitioner solely, the information would be lacking. Hence, loss to endpoint findings is considered to be negligible.
Conclusions
In this pooled analysis of 78,546 individuals, self-reported alcohol intake of 1 to 20 drinks/week was associated with reduced risk of any stroke. The risk of any stroke if anything tended to be increased among individuals with heavy alcohol intake, indicating a j-shaped association. However, no significant association was found between high alcohol consumption and risk of any stroke. The pattern was similar for ischemic stroke and haemorrhagic stroke, but only statistically significant for ischemic stroke. Finally, we found no evidence to support a causal relation of a linear association between alcohol intake and total stroke from genetic estimates. However, this could be explained by lack of power in the MR analysis.