Introduction
Acute ischemic stroke (AIS) is a major cause of mortality and long-term disability world-wide that lacks curative therapies [1,2]. Recent key advancements in pharmacological thrombolysis and mechanical endovascular thrombectomy remain limited by a narrow therapeutic window and restrictive eligibility [3,4]. Additionally, the spectrum of rehabilitation paradigms implores evidence base [5], while efforts targeting neuroprotection largely fail to translate to human therapies [6]. AIS research has historically focused on these central nervous system (CNS) interventions to reduce infarct volume and address neuronal viability. More recently, however, detrimental effects of AIS on the heart [7] and immune system [8] have gained popularity. Indeed, medical complications following AIS such as pneumonia are strong predictors of mortality and functional outcome [9,10]. To that end, we queried the National Center for Biotechnology Information and National Library of Medicine database through PubMed using a combination of keywords including “ischemic stroke” and the system of interest to identify relevant literature addressing systemic effects of AIS. We found a small but growing body of evidence pointing to far-reaching pathophysiological consequences in peripheral tissues including immune, respiratory, urinary, cardiovascular, gastrointestinal, musculoskeletal, and endocrine systems (Supplementary Figure 1). Following AIS, the majority of these organ systems suffer disturbances ranging in severity from subclinical laboratory abnormalities to life-threatening arrhythmias or infections (Figure 1). In this current review, we discuss the mechanisms by which AIS induces these systemic pathophysiological responses and summarize their subsequent clinical manifestations.
The crosstalk between the strokeaffected brain and end organ systems
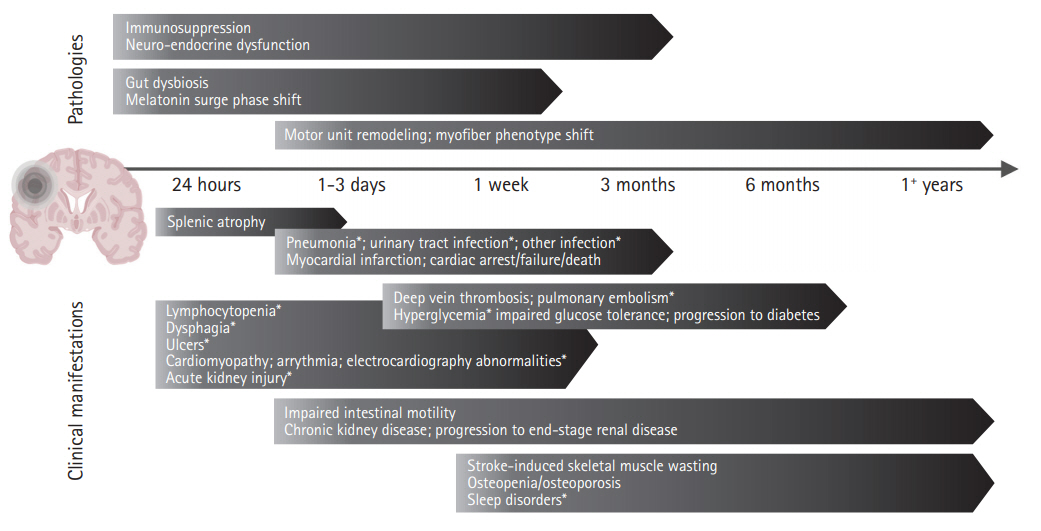
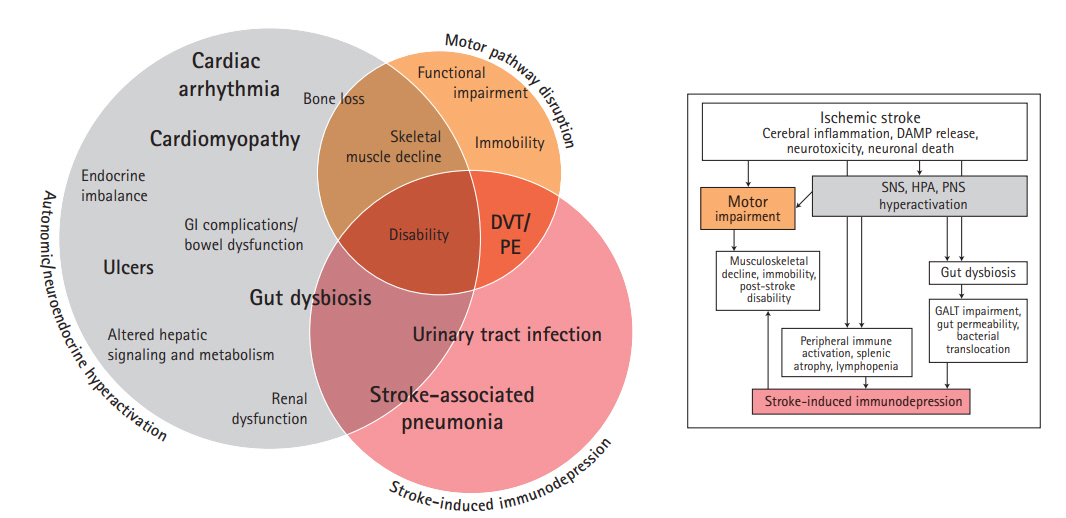
It is now widely accepted that AIS has dramatic consequences on the fine balance between the brain and the rest of the human body (Table 1, Supplementary Table 1) [9,11]. Following AIS, many systems demonstrate time-dependent progression of acute stroke-induced alterations preceding chronic deficits (Figure 2) [12-14]. These alterations are believed to be mediated by three overlapping mechanisms involving immune, autonomic, and motor pathways (Figure 3).
Shortly after AIS, a severe state of immunodepression is seen resulting in the high incidence of post-stroke infections [9,11]. This post-stroke immunodepression is mediated by the autonomic and hypothalamic pituitary adrenal axis (HPA) activation and the interaction of the stroke-affected brain with the immune system through complement activation and release of damage-associated molecular patterns (DAMPs) [15,16]. Additionally, HPA activation is thought to be a critical channel through which multiple other systems are dysregulated, resulting in cardiac, renal, and gastrointestinal imbalances. Furthermore, motor pathway impairment secondary to AIS contributes to muscle wasting while the associated immobility increases the risk of deep vein thrombosis (DVT) and pulmonary embolism (PE) (Table 1). These factors overlap resulting in the worsening morbidity and mortality of AIS (Figure 3). In the following sections, these overlapping mechanisms and their subsequent clinical implications are discussed in further detail.
Stroke-induced immunodepression
Within hours of AIS, autonomic activation and release of DAMPs affect the various immune cells in the body. Immune cells from lymphoid populations in the spleen, gut-associated lymphoid tissue (GALT), and bone marrow reach brain vasculature and parenchyma [17,18]. Together with resident microglia, the arriving neutrophils, monocyte/macrophages, and innate lymphocytes (e.g., natural killer [NK] cells) respond by producing proinflammatory mediators, after which T- and B-cell activation delivers its adaptive response [17-19]. The roles of T-cell subsets have been thoroughly investigated, where pro-inflammatory T-helper (Th1), Th17, γδ T-cells, and cluster of differentiation 8+ (CD8+) T-cells promote initial tissue damage, enhance blood brain barrier breakdown, and contribute to neuronal apoptosis [19]. Following this acute activation phase, an abrupt anti-inflammatory shift supervenes. This process is thought to be mediated by autonomic and neuroendocrine dysfunction, monocyte deactivation, and NK cell impairment that suppress immune activity. In addition, peripheral mobilization and apoptosis decrease blood lymphocyte counts by half [20]. The shift toward anti-inflammatory Th2 cells and expansion of protective forkhead box protein P3+ (FoxP3+) regulatory T-cells (Tregs) protect against further neuroinflammation [21]. While limiting excessive immunological brain injury, the diminished immune capacity prevents sufficient inflammatory response to infection [15]. Though exact mechanisms of prompt immune activation and opposing immunodepression are not fully understood, known participants in brain-immune communication after stroke are described below.
DAMPs and endotoxin tolerance
DAMPs are intracellular biomolecules that initiate an immune response upon release from necrotic cells. Upon detection by pattern recognition receptors on immune cells (e.g., receptor for advanced glycation end products [RAGE], toll-like receptors [TLRs]), DAMPs acutely amplify pro-inflammatory cytokine production [15]. This disproportionately large immune reaction incites immature monocyte migration from bone marrow, proliferation of bone marrow-derived suppressor cells in the spleen, lymphopenia, and immune cell apoptosis [22,23]. DAMP-induced exhaustion of monocyte function yields a state of endotoxin tolerance, a phenomenon that protects the stroke-affected brain from further damage but leaves immune cells unable to elicit adequate pro-inflammatory reaction to insult [8].
Monocytes from AIS patients exhibit decreased surface expression of major histocompatibility complex (MHC) class II molecule human leukocyte antigen D related (HLA-DR) and consequently exist in a state of diminished antigen presentation [24]. Neuronal chromatin-associated nuclear protein high-mobility group box 1 protein (HMGB1), a pivotal DAMP in stroke pathology, is elevated in serum within hours [15]. Both markers of immunodepression, low MHC II and HLA-DR at post-stroke day one predicted infection up to 2 weeks later [24], while preclinical blocking of the HMGB1-RAGE pathway attenuated immunodepression and lymphocyte inactivation [22]. Mitochondrial DAMPs are also markedly increased in plasma after stroke. Circulating mitochondrial DNA strongly correlated with the impaired response of refractory-state monocytes and infection [8]. Furthermore, healthy monocytes cultured in stroke patient serum containing mitochondrial DNA exhibited the same refractory state [8].
Invariant natural killer T (iNKT) cells are a unique group of innate immune cells that survey the blood for circulating immunogens. Recent data suggest iNKT cells in liver sinusoids detect and respond to brain injury [25]. Following middle cerebral artery occlusion (MCAO), iNKT cells demonstrated decreased crawling activity specific to cerebral ischemia, as hindlimb ischemia-reperfusion had no effect on iNKT mobility [25]. Post-stroke changes to iNKT behavior also contribute to immunodepression and increased infection risk. While iNKT numbers were not altered by stroke-induced lymphopenia, activation marker CD69 increased in peripheral blood and liver iNKT populations after MCAO prior to the anti-inflammatory cytokine shift. Neutrophil infiltration, edema, and bacterial load also increased in the lungs within 24 hours, consistent with clinical outcomes [25].
Splenic volume and cell cycling
The spleen is a lymphoid organ functioning in blood filtration and immune response. Preclinical stroke studies describe immediate decreases in splenic volume and T-cell antigen response [20,23]. Similarly, stroke survivors experience splenic atrophy with re-expansion beginning after 48 hours [12]. Prompted by sympathetic hyperactivation, contributors to splenic contraction include immune cell release followed by apoptosis of lymphocyte subsets: B-, T-, and NK cells [16,20,26]. Confirming the impact of apoptosis in immunodepression, experimental treatment with systemic caspase inhibitor quinolyl-valyl-O-methyl-aspartyl-[-2,6-difluorophenoxy]-methyl ketone (Q-VD-OPH) minimized stroke brain injury, improved splenocyte survival, and reduced bacteremia [27].
To delineate the spleen’s role in stroke injury, independent preclinical studies showed splenectomy either 2 weeks before MCAO [28] or in the immediate hours afterward [29] decreased infarct volume and neurological deficits. Interestingly, splenectomy immediately before stroke induction did not improve infarct volume or neuroimmune response [17,30], though variance in stroke model, post-operative recovery, and immune compensation warrant further investigation [10,30].
Gut dysbiosis
The microbial population in the intestinal tract interacts with specialized GALT to regulate T-cell homeostasis and establish the maturing immune system. Disruption of the intestinal microbiome, or gut dysbiosis, is observed following AIS [14]. Because microbial metabolites influence immune polarization, gut dysbiosis affects intestinal lymphocyte populations [31]. Due to its role in immune response and systemic inflammation, gut dysbiosis is addressed in the context of neuroimmune dysregulation.
In response to inflammatory stress and altered vagal communication with the intestinal tract, gut dysbiosis after stroke is characterized by a reduction in species diversity, imbalance in phyla predominance, and altered metabolite production [14,32]. Preclinically, dysbiosis directly primed inflammatory γδ T-cells in the gut, which then trafficked to the brain and exacerbated stroke evolution [31]. Recolonizing germ-free mice with dysbiotic bacteria from stroke-affected mice yielded larger lesions and deficits after MCAO, while healthy fecal transplantation normalized microbiota, reduced infarct size, and attenuated proinflammatory T-cell polarization [14]. Following the post-stroke immunosuppressive shift, mesenteric lymph node dendritic cells prompt migration of protective Tregs to the gut to decrease γδ T-cell movement to the brain [19]. Compared to mice with less severe dysbiosis, mice with excessive dysbiosis had significantly more γδ T-cells, fewer Tregs, larger infarcts, and greater neurological deficits [33].
Clinically, AIS patients presented with altered fecal bacterial counts and organic acid levels, as well as disrupted biomarkers of metabolism and inflammation [32]. Gut microflora are further disturbed by antibiotic administration for infection control [34]. Intestinal edema and inflammation after experimental stroke compromised mucosal barrier function and may enable bacterial translocation to various organs [35]. Indeed, bacteremia occurred acutely following rodent stroke [34] and was observed in AIS patients at admission [14]. Microbes detected in the lungs originated in the host’s small intestine, with translocation to the spleen, liver, and mesenteric lymph nodes as early as 24 hours [34,35]. These findings indicate gut dysbiosis is both a consequence of and subsequent contributor to stroke pathophysiology, though a clinical link between dysbiosis and stroke outcomes remains to be established.
The systemic consequences of stroke-induced immunodepression
The critical suppression of the immune system manifests clinically as infection, including stroke-associated pneumonia (SAP) and urinary tract infection (UTI), the most frequently encountered complications in stroke patients [36].
Stroke-associated pneumonia
Murine studies report increased bacterial load in lungs and peripheral blood by 24 hours post-MCAO [20]. Obstructing sympathetic activity with a β-blocker, however, prevented bacterial infection and reduced mortality highlighting the role of sympathetic system activation [20,37]. Immunodepression was further demonstrated using Streptococcus pneumoniae exposure: while 200,000 colony-forming units (CFUs) were required to cause pneumonia in sham animals, only 200 CFUs were necessary in stroke mice, again preventable with sympathetic blockade [37]. Marked lymphopenia was prevented through glucocorticoid receptor blockage, while both HPA and sympathetic inhibition protected against lymphocyte apoptosis [18].
Stroke-induced parasympathetic stimulation is widely accepted as another contributor to infection through initiation of anti-inflammatory pathways [14,38,39]. Vagal signaling and cholinergic activation target α7-nicotinic acetylcholine receptors (α7-nAChR) across cell types, including lung epithelial cells and resident immune alveolar macrophages [40]. Mice after MCAO presented with significant parasympathetic response and pneumonia, while vagotomy or α7-nAChR-deficiency maintained pulmonary immune defense and prevented SAP [40].
Clinically, SAP is a major complication of AIS affecting onethird of patients [41]. In contrast to preclinical data, human studies utilizing β-blockers to address infection report mixed outcomes, from protective [42] to unfavorable [43]. Aspiration is another important mechanism of SAP [9], though prevention measures alone do not eliminate infection. Indeed, a multi-center study established dysphagia and immunodepression as independent predictors of SAP [24]. Dysphagia correlated with SAP only in patients with low monocytic HLA-DR, suggesting screening for both dysphagia and immunosuppression could identify SAP risk [24]. Though SAP is linked to functional decline and mortality [36], prophylactic administration of antibiotics failed to improve either outcome in clinical trials [44,45]. This emphasizes the knowledge gap regarding complex pathophysiological mechanisms of post-stroke immunodepression and the fragile lung-brain axis.
Urinary tract infection
AIS severity is an independent predictor of UTI [41] which is in turn associated with worse outcomes, longer hospital stays, and poor discharge outcome [41]. Despite lowering UTI frequency, prophylactic antibiotics did not benefit functional outcome or reduce mortality in clinical trials similar to SAP [46]. Risk factors include catheter use, incontinence, and post-void residual urine volume [47], though high rates of UTI have been reported in patient cohorts both with (50%) and without (24%) indwelling catheters [48]. Programs that prompted routine assessment, stop orders, or early removal reduced catheter duration and incidence of UTI [49], as did measuring post-void urine volume with portable bladder ultrasound [47]. Antimicrobial or antibiotic-impregnated catheters have been shown to delay or prevent bacteriuria, but evidence of UTI reduction is scarce [50] and highlights immune system complexity after stroke.
Autonomic and neuroendocrine dysregulation
Dysregulation of autonomic and HPA systems has widespread effects on all body organs (Figure 3). The mechanisms by which the autonomic system communicates with the peripheral immune system have been most thoroughly investigated and will be discussed in depth first.
Sympathetic hyperactivation
The pro-inflammatory cytokine surge after AIS activates the sympathetic nervous system (SNS), triggering catecholamine production at the adrenal medulla and sympathetic nerve terminals [20]. This initiates immune cells mobilization, stimulates their apoptosis, and inhibits further cytokine production [20]. Murine studies identified sympathetic activation as the direct cause of spontaneous bacterial infection after stroke [20], while pharmacological inhibition of splenic adrenergic receptors reduced infarct volume, preserved splenic weight, and protected against high bacterial load [51]. Though splenectomy was protective, denervation alone did not alter splenic weight or infarct volume [51] suggesting a regulatory role for circulating catecholamines over direct sympathetic innervation. Indeed, patients with post-stroke infection presented first with increased catecholamine levels [52].
On the other hand, noradrenergic-mediated innervation, rather than humoral input, is responsible for post-stroke iNKT behavior. Direct injection of norepinephrine into livers of shamoperated mice mimicked the immunosuppressive iNKT response to stroke and increased infection risk, whereas β-adrenergic receptor inhibition or reduction of hepatic noradrenergic nerve terminals attenuated the MCAO-induced immunosuppressive shift in mice [25], similar to results in the spleen [51].
HPA axis dysregulation
HPA axis overactivation releases glucocorticoids from the adrenal cortex, promoting the immunosuppressive shift to anti-inflammatory cytokine production [20]. Glucocorticoids have been shown to impede pro-inflammatory cytokine production as well, and stunt immune cell proliferation to promote apoptosis [53]. Clinically, AIS prompts an abrupt increase in cortisol, which is linked to post-stroke infection, mortality, and functional dependence [54]. Experimental blocking of glucocorticoid receptors prevented the immunosuppressive stroke-associated lymphopenia [18,20]. Many studies report diurnal pattern disruption of cortisol after stroke, though full implications of the shift remain unknown [54].
Parasympathetic vagal activation
The parasympathetic nervous system is involved with neuroimmune regulation following stroke through inflammatory stimulation of the vagus nerve [40]. The subsequent release of acetylcholine activates α7-nAChR, triggering the vagal cholinergic anti-inflammatory pathway [55] which inhibits pro-inflammatory macrophage activity. Studies employing vagal stimulation [38] and α7-nAChR agonists [39] confirm vagal/α7-nAChR involvement in reducing cerebral inflammation. The other consequence of the parasympathetic vagal activation is its effect on the brain and peripheral NK cell regional population in mice and patients [26]. A decrease-then-recovery of NK counts occurred in the periphery, but the opposite—initial increase, then subsequent contraction—was observed in the brain. In addition, cholinergic exposure suppressed NK function in the brain but not in the periphery [26]. This suggests compartment-specific mechanisms may be ideal targets for preventing immunodepression in the periphery while leaving the protective shift in the brain undisturbed.
The systemic consequences of autonomic/neuroendocrine dysregulation
Overstimulation of sympathetic, HPA, and parasympathetic pathways mediates the systemic disease progression, prompting a clinical cascade of cardiac, gastrointestinal, hepatic, renal, and endocrine complications.
Cardiovascular dysfunction
Stroke severity, disability, and mortality are higher in patients with lower cardiac function [56,57]. Conversely, AIS induces cardiac dysfunction without prior risk factors or pre-existing heart disease [57,58]. This manifests with electrocardiographic abnormalities, elevated serum cardiac troponin T in the absence of myocardial infarction, depressed cardiac function in the acute phase, or severe complications such as arrhythmias or cardiac arrest [56-58]. Incidentally, 19% of AIS patients had a serious or fatal cardiac event in the first 3 months [10].
Takotsubo cardiomyopathy (TTC) is a stress-induced transient weakening and ballooning of the left ventricle that is seen in a subset of AIS patients. Commonly asymptomatic, TTC may mimic myocardial infarction in injury biomarkers and electrocardiography and is associated with insular infarcts and poor outcomes [59,60]. Heart function typically returns to normal over several weeks, but severe cases increase risk for cardiac embolism, respiratory failure, and death [59].
Cardiac dysfunction following AIS has been attributed to sympathetic and parasympathetic imbalance. One study confirmed overstimulation of cardiac sympathetic innervation using the catecholamine synthesis rate-limiting enzyme tyrosine hydrolase [7]. Sympathetic hyperstimulation also blunts vagal influence, as confirmed in stroke patients via heart rate variability testing [61]. HPA activation and hypothalamic paraventricular nucleus output also prompted cardiac arrhythmias and impaired cardiac output in rat models [62]. Brain regions of cardiovascular interest are the brainstem (e.g., rostral ventrolateral medulla) and insular cortex. The laterality hypothesis maintains left and right insular cortices control cardiac parasympathetic and sympathetic activity respectively, and while patient studies indicate heightened severity of cardiac dysfunction with insular strokes, presentation and degree of laterality remain controversial [63,64].
Gastrointestinal and metabolic impairments
Autonomic dysfunction impairs enteric communication and, consequently, gastrointestinal function. Additional contributors include inactivity, diet, and various medications [65]. Preclinical and clinical studies alike report bowel dysfunction and gastrointestinal impairment after stroke [14]. Reduced intestinal motility results in constipation and new-onset fecal incontinence in up to half of AIS patients [65]. Rodent studies identified histological damage, gastric edema, hyperemia, altered cellular composition, and hemorrhagic erosion of the gastric mucosa after stroke [66], which present clinically as ulcers [67]. Acid-suppressive therapy may reduce gastrointestinal bleeding [67], but the underlying intestinal inflammation, gut dysbiosis, and autonomic contributors remain.
Metabolic homeostasis in the liver is impaired after AIS and correlates with infarct volume [68]. Stroke is associated with reduced unconjugated bilirubin and hemoglobin levels at admission [68] while increasing glutamate-metabolizing enzymes associated with liver injury (e.g., glutamate oxaloacetate transaminase [GOT]), a possible prompt for peripheral glutamate metabolism in response to glutamate release from the ischemic brain [68]. Plasma and neuronal sources of GOT support scavenging [69] and excitotoxic glutamate metabolism [70] after AIS respectively, though post-stroke implications of hepatic GOT are yet to be revealed. Additionally, experimental stroke promoted inflammation, DNA fragmentation, and apoptotic signaling in the liver through kinase activation (i.e., c-JUN N-terminal kinases [JNKs], extracellular signal-regulated kinases [ERKs]) [71]. Compromised hepatic insulin signaling, increased expression of gluconeogenic genes, and increased endoplasmic reticulum stress have been reported in the liver and are attributed to the post-stroke catecholamine surge [72].
Hepatic ketogenesis is another hepatic disturbance seen in animal models. Stroke-affected mice develop ketogenesis when fed fat-rich diet, even when the diet was not sufficient to trigger ketosis pre-stroke [73]. This phenomenon may have a potential role in angiogenesis and neuroprotection following AIS [74].
Kidney dysfunction
Stroke-induced autonomic hyperactivation brings an increase in sympathetic activity and systemic inflammation, disrupting renal homeostasis [75]. Up to one-third of stroke patients experience renal impairment during hospital stay [76]. AIS patients can develop acute kidney injury, defined as decreased urine output or increased absolute serum creatinine within 48 hours [77]. Influential factors beyond sympathetic activity include hydration status, contrast nephrotoxicity, and vascular intervention [77]. Stroke survivors can also suffer progression of chronic kidney disease that can evolve to end stage renal disease [78]. Post-stroke renal dysfunction upon hospitalization is a prognostic indicator of mortality at 10 years [76] and thus supports the case for identification and management.
Other disturbances include urinary incontinence, with or without urgency, in half of AIS patients. Given the complex neurological control over micturition, lesion size may be of greater concern than specific localization to certain brain areas [79].
Endocrine imbalance
Stroke-induced sympathetic signaling to the pancreas blocks insulin release, making hyperglycemia common after stroke even in non-diabetic patients [80]. Hyperglycemia commonly extends beyond the acute period to involve impaired glucose tolerance at discharge or progression to diabetes [80]. While hyperglycemia is associated with larger infarcts and worse functional outcome, clinical trials found no therapeutic benefit to intensive blood glucose control, noting induced hypoglycemia is of equal concern [81].
Low levels of thyroid hormone triiodothyronine (T3) have been observed immediately following stroke in more than half of patients, independent of pre-existing thyroid conditions [82]. Low serum T3 and positive thyroid autoantibodies were associated with worse outcomes following AIS [82,83]. Experimental T3 administration after MCAO reduced cerebral edema by inhibiting aquaporin-4 (AQP4) expression [84], but clinical T3 supplementation has not been tested. Possible explanations of thyroid hormone disruption after AIS include pro-inflammatory mediators and glucocorticoid influence [82].
Stroke disrupts the circadian rhythms and melatonin production, even when lesions do not affect pineal gland control [85]. A preclinical study observed a phase shift in melatonin release after stroke [85], and clinical AIS studies reported decreased melatonin at day 1 [86]. Melatonin treatment was neuroprotective after rodent stroke, where proposed mechanisms are mitochondrial apoptosis pathway inhibition and promotion of neuronal survival pathways [87], suggesting the melatonin decrease following stroke may be from swift metabolism to combat stroke-induced damage.
Motor pathway disruption
Focal weakness as a result of motor pathway disruption is commonly seen post-AIS [1,2,5]. In addition to the motor system lesion, the pathophysiology of motor impairment stems also from the neuroplastic response, which alters upstream/downstream non-lesioned networks [88]. Neuroimaging advancements (e.g., functional magnetic resonance imaging) foster detailed study of motor mapping after stroke [89]. Stroke patient testing found functional coupling between cortical activity and muscle output is markedly reduced in the acute phase and remains as such even when motor performance improves [90]. Interestingly, patients without apparent improvement still exhibited complex changes over time due to motor network connectivity [91]. Investigated mechanisms of motor functional recovery include axonal sprouting [92,93], white matter repair [94], neurogenesis [95], and ipsilesional/contralesional network reorganization [96]. Just as consequences of stroke disability maladaptively alter cortical mapping and hinder recovery [97], interventions targeting beneficial neuroplastic reorganization can strengthen tracts and improve recovery [98,99].
Systemic consequences of motor pathway disruption and immobility
Motor pathway disruption and secondary immobility result in increased risk of venous thromboembolism (VTE) formation and widespread impacts to the musculoskeletal system.
Deep vein thrombosis and pulmonary embolism
Post-stroke VTE risk ranges from 8% to 30% and is independently associated with AIS severity [1,100,101]. Especially in the acute phase when hemodynamics, inflammation, and immobility compound, DVT may develop and travel to the lungs, yielding life-threatening PE [100]. Atherosclerotic predisposition was explored as a potential link between AIS and VTE, but causation was determined unlikely [101]. Infection, however, is associated with both coagulation system activation and immobilization [101]. As such, stroke-associated infection, prothrombotic activity, inflammation, dehydration, and immobility all serve roles in VTE development after stroke [101]. Prophylactic chemical DVT dosages and intermittent pneumatic compression reduce DVTs in AIS patients [4,102].
Bone loss and remodeling disorder
Chronic stroke patients experience bone loss as mineral density declines significantly within one year and fracture risk increases up to 7-fold [103]. The pathophysiology of bone loss extends beyond immobility and asymmetric weight-bearing to disruption of central brain centers controlling skeletal homeostasis [104]. Inner ear vestibular lesioning in rats decreased bone formation without affecting resorption or locomotor activity [104]; these alterations in bone metabolism were attributed to SNS outflow and prevented with β2-adrenergic receptor blockade [104]. Likewise, serum bone formation marker N-terminal propeptide of type 1 procollagen (PINP) decreased independent of activity level in MCAO rats, with no change in resorption marker C-terminal telopeptide of type I collagen (CTX) [105]. Further investigation is needed to define exact pathways involved with brain control of bone metabolism and stroke-induced remodeling disorders.
Skeletal muscle decline
Stroke prompts severe alterations to skeletal muscle tissue. Myofiber analysis identified a shift in myosin heavy chain predominance toward fast-twitch isoforms with initial denervation that declined as motor recovery and spasticity progressed [106]. Reduced gait speed is also associated with the poststroke atrophy response [106]. Catabolic activity in mouse strokeaffected muscle was measured as increased expression of apoptotic and proteasome proteolytic markers and significantly correlated with infarct size [107]. Proposed contributors to skeletal muscle decline after stroke include reduced feeding and inactivity, sympathetic activation, and infection. Weight loss and muscle wasting, however, were not altered by high-caloric diet, sympathetic blockade, or antibiotic treatment [107], suggesting mechanisms unique to stroke are responsible for the observed pathophysiology.
Exact molecular mechanisms of AIS-induced muscular decline are unclear. Myostatin is upregulated in post-stroke paretic muscle [13,108]. Myostatin inhibits the anabolic Akt/mammalian target of rapamycin (mTOR) pathway while increasing ubiquitin-proteasome activity and autophagy-lysosome proteolysis [108]. Inhibiting myostatin with PINTA745 promoted recovery of muscle mass and function in rodents [108]. Providing similar results, progressive resistance training decreased patient myostatin mRNA and stimulated muscle hypertrophy [109]. Separately, stroke upregulated pro-inflammatory interferon-γ-induced protein 10 (IP-10/CXCL10) in rat paretic muscle while prompting significant loss of brain-derived neurotrophic factor (BDNF), an important mediator for muscular repair [13].
Data propose the systemic inflammatory response following AIS reaches skeletal muscle and influences its reparative capacity. Clinical data regarding muscle pathophysiology and associated mechanisms are limited and identifies a need for evidence-based models of rehabilitation that address stroke-induced skeletal muscle decline.
Conclusions and future directions
A growing body of evidence recognizes acute and chronic repercussions in peripheral tissues that worsen stroke evolution and alter prognosis (Figures 1 and 4). The aforementioned ramifications of ischemic stroke on autonomic, neuroendocrine, immune, and motor systems are a conduit for injury response to move beyond the CNS. As presented herein, the individual clinical manifestations of AIS cannot simply be attributed to a single pathophysiological mechanism. Rather, each complication stems from a myriad of influencers and, likewise, prompts new deficits while magnifying the existing disease state.
The disappointing translational failure of AIS therapeutics and neuroprotective agents from bench to bedside is markedly pronounced in comparison to clinical trial successes across various other medical conditions [110]. This is partly related to the widespread complex pathophysiology of AIS in the body. Central to this issue is a call for integrative progression in stroke research, where experts consolidate therapeutic knowledge to address the complexity of stroke pathophysiology.
The concept of AIS as a systemic disease is clear when considering how pathophysiological mechanisms reach nearly every organ system in the human body. Hence, reductionist approaches that fail to consider bidirectional crosstalk between the CNS and periphery limit our ability to conceptualize new paradigms of patient care. A comprehensive understanding of post-stroke systems biology offers new direction to mechanistic discovery, defining multi-system targets to include the SNS, gut microbiome, neuroimmune interactions, and musculoskeletal metabolism. As such, collaborative research efforts across clinical specialties are necessary to inform methods and achieve the future of integrative stroke therapeutics.