Development and Testing of Thrombolytics in Stroke
Article information

Abstract
Despite recent advances in recanalization therapy, mechanical thrombectomy will never be a treatment for every ischemic stroke because access to mechanical thrombectomy is still limited in many countries. Moreover, many ischemic strokes are caused by occlusion of cerebral arteries that cannot be reached by intra-arterial catheters. Reperfusion using thrombolytic agents will therefore remain an important therapy for hyperacute ischemic stroke. However, thrombolytic drugs have shown limited efficacy and notable hemorrhagic complication rates, leaving room for improvement. A comprehensive understanding of basic and clinical research pipelines as well as the current status of thrombolytic therapy will help facilitate the development of new thrombolytics. Compared with alteplase, an ideal thrombolytic agent is expected to provide faster reperfusion in more patients; prevent re-occlusions; have higher fibrin specificity for selective activation of clot-bound plasminogen to decrease bleeding complications; be retained in the blood for a longer time to minimize dosage and allow administration as a single bolus; be more resistant to inhibitors; and be less antigenic for repetitive usage. Here, we review the currently available thrombolytics, strategies for the development of new clot-dissolving substances, and the assessment of thrombolytic efficacies in vitro and in vivo.
Introduction
Stroke is one of the leading causes of death and disability worldwide [1]. It is also one of the most frequent causes of dementia and epilepsy. Moreover, the incidence of stroke will be increasing because of population aging [1]. The impact of stroke on modern societies is and will be enormous because of loss of lives, loss of quality of life of surviving patients and their families, and its huge socioeconomic burden.
Until 2014, the only evidence-based therapy to reverse the neurological deficit in acute ischemic stroke was the administration of intravenous tissue plasminogen activator (tPA) within 4.5 hours from symptom onset [2]. Since 2015, several studies have demonstrated the substantial benefits of mechanical thrombectomy in patients with acute stroke due to large vessel occlusion [3-5]. Despite the advances in recanalization therapy, approximately half of patients with severe ischemic stroke remain disabled or die [3-5]. Moreover, access to mechanical thrombectomy is quite limited in many countries owing to lack of resources or experts [6]. Furthermore, mechanical thrombectomy will never be a treatment for every ischemic stroke because many of them are caused by occlusion of cerebral arteries that cannot be reached by intra-arterial catheters.
To address these needs, various non-interventional treatments are being developed, as described recently [7]. Thrombolytics, compared with other treatments, appear to hold greater promise for stroke management since they have already demonstrated clinically proven therapeutic utility. However, thrombolytics have shown relatively limited efficacy and notable hemorrhagic complication rates, leaving room for improvement. In this article, we summarize the currently available thrombolytics, including their advantages and shortcomings, and describe strategies for developing and testing principally new thrombolytics in pre-clinical and clinical settings. A comprehensive understanding of basic and clinical research pipelines and the current status of thrombolytic therapy will help facilitate the development of new thrombolytics.
Current thrombolytics
Tissue plasminogen activator
Molecular structure of tPA
tPA was discovered in animal tissues in 1947 and successfully purified in 1979 [8]. tPA is synthesized as a single-chain protein, but can be converted into a two-chain form by plasmin or kallikreins without loss of fibrinolytic efficiency [9]. It is secreted by vascular endothelial cells, glial cells, and neurons [10-12]. tPA consists of five functional domains (Figure 1): (1) a fibronectin-like or finger domain (F, 4–50 amino acids [AA]), (2) an epidermal growth factor (EGF)-like domain (E, 50–87 AA), (3) a kringle 1 domain (K1, 87–176 AA), (4) a kringle 2 domain (K2, 176–256 AA), and (5) a catalytic serine protease domain (P, 276–527 AA) [13]. tPA binds to fibrin through its F, K2, and probably, K1 domains, while its F and E domains are responsible for a fast clearance by hepatocytes [14,15]. The average half-life of tPA in plasma is approximately 6 minutes [16]. Carbohydrate chains, which are connected to tPA via three N-glycosylation sites at residues 117, 184, and 448, and one O-fucosylation site at Thr61, influence the clearance efficiency and plasma half-life [17,18].
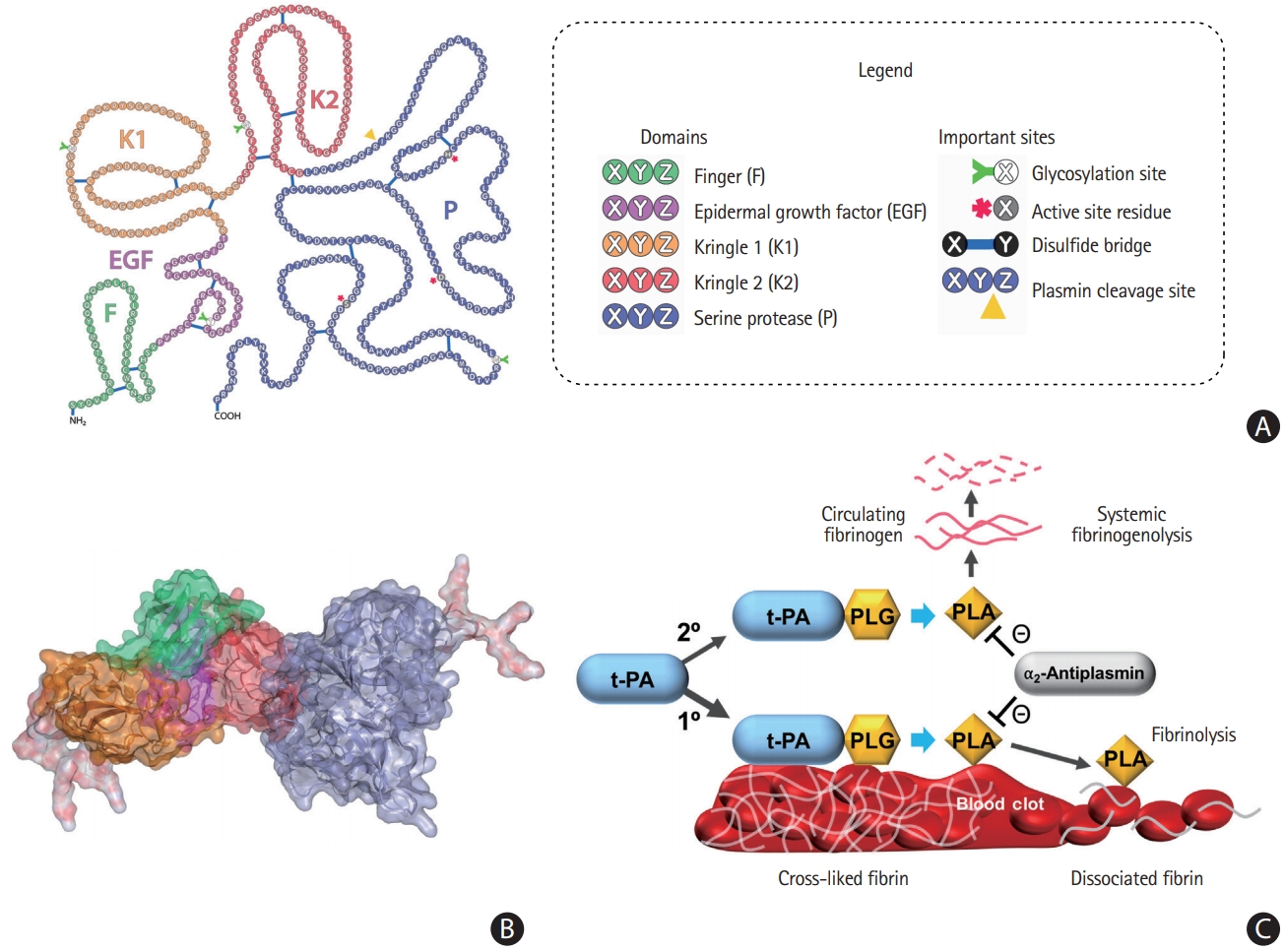
Molecular structure and fibrinolytic function of tissue plasminogen activator (tPA). (A) The F domain (green) is involved in binding to fibrin, which stimulates the activity of tPA. Binding of F and epidermal growth factor (EGF)-domain (violet) to the low-density lipoprotein receptor-related protein 1 (LRP1) receptor on the surface of a hepatocyte is involved in the clearance of tPA. The K1 domain (orange) is homologous to the K domains of urokinase and desmoteplase, and glycosylation at Asn117 on the K1 domain influences tPA uptake in the liver [34]. The K2 domain (red) has a lysine-binding site, which binds to partially degraded fibrin and other proteins containing C-terminal lysines. This could be the basis of enhancement of plasminogen activating activity of tPA by interacting with cofactors other than fibrin. The trypsin-like serine protease P domain (steelblue) is responsible for the catalytic activity of tPA. Upon tPA binding to fibrin, the catalytic activity is increased by both colocalization of plasminogen and conformational change of tPA into a more active state. (B) Carbohydrate chains (white+red) affect the half-life of tPA by interacting with the mannose receptor and asialoglycoprotein receptor on the endothelial cells in the liver [33,35]. In addition, carbohydrate chains influence the catalytic activity by preventing interactions between the domains of tPA [35]. (C) The cartoon in the lower right panel illustrates the mechanism of tPA-mediated fibrinolysis in thrombi and circulating blood. Fibrinolysis (1º) occurs after tPA binds to fibrin-associated plasminogen (PLG) on the surface of the clot by generating plasmin (PLA), and thereby breaking the cross-links between fibrin molecules. PLA could also break down circulating fibrinogen (2º), and thus potentially causing bleeding complications. Plasmin activity is inhibited by α2-antiplasmin. Images of molecular structures of tPA that were adapted from Mican et al. [36], with permission from the Elsevier.
Biological function of tPA
tPA is a serine protease that converts plasminogen into active plasmin, which can degrade fibrin clots [19]. Moreover, it also regulates many other physiological processes, such as neuronal migration, neurite outgrowth, glutamatergic neurotransmission, long-term potentiation, synaptic plasticity, and neurovascular coupling [20-26].
Clinical use of tPA
Clinical research has mostly revolved around alteplase, a recombinant form of tPA, which is still the only Food and Drug Administration (FDA)-approved thrombolytic drug for the treatment of acute ischemic stroke. It was approved by the FDA for the management of acute myocardial infarction in 1987, acute massive pulmonary embolism in 1990, and acute ischemic stroke in 1996 [27]. In a meta-analysis of individual patient data from 6,756 patients in nine randomized trials comparing with a placebo or open control, alteplase significantly increased the odds of a good outcome when given within 4.5 hours after ischemic stroke. There was an absolute increase of approximately 2% in the risk of early death caused by intracranial hemorrhage, although it was offset by an increase in disability-free survival. The rates of alteplase-mediated recanalization can be as low as 5% to 10% for proximal large vessel occlusion, although they are much higher for more distal occlusions [28].
Protein engineering of alteplase
Many attempts have been made to improve biochemical properties of alteplase by protein engineering to construct different variants, for example, monteplase (with a mutation Cys84Ser in the EGF domain) [29], lanoteplase (with a mutation Asn117Gln in the K1 domain) [30], pamiteplase (with K1 domain deleted and a mutation Arg275Glu) [31], and amediplase (with K2 domain from tPA and P domain from urokinase) [32]. A detailed description of the domains is presented in Figure 1 [33-36]. All these drugs have prolonged plasma half-life but decreased fibrin specificities. In contrast, tenecteplase (TNK), which is generated by a few point mutations, has a longer plasma half-life and a 14-fold higher fibrin specificity owing to the substitution of Thr103Asn (introducing glycosylation site), Asn117Gln (deleting glycosylation site), and Lys296-His297-Arg298-Arg299 with four alanines [37]. The latter substitutions decrease plasminogen activator inhibitor 1 (PAI-1) inhibition up to 80-fold [37]. Clinical trials with TNK have revealed a decrease in bleeding complications [38].
Urokinase
Urokinase was discovered in human urine in 1947 [39]. It is produced by vascular endothelial cells, smooth muscle cells, epithelial cells, fibroblasts, monocytes/macrophages, and various types of cancer cells [40-44]. The enzyme is involved in a variety of physiological processes, such as cell migration, tissue remodeling, complement activation, pro-hormone conversion, and wound healing [45]. Urokinase is a serine protease, which consists of three domains: (1) an EGF-like domain (E, 1–49 AA), (2) a kringle domain (K, 50–131 AA); and (3) a catalytic serine protease domain (P, 159–411 AA) with a catalytic triad His204, Asp255, and Ser356 [46]. Urokinase is cheaper than alteplase and widely used for the treatment of ischemic stroke in developing countries [47]. In developed countries, urokinase is clinically used for treating deep venous thrombosis and peripheral arterial occlusive disease. Urokinase was approved by the FDA for the systemic treatment of pulmonary embolism [48] and catheter-directed treatment of acute myocardial infarction [49,50].
Streptokinase
Streptokinase was discovered in β-hemolytic streptococci in 1933 [51]. This indirect plasminogen activator is naturally involved in the propagation of streptococcal infections via the breakdown of restraining fibrin barriers [52]. It is approved by the FDA for acute myocardial infarction, pulmonary embolism, deep vein thrombosis, and arterial thrombosis treatments.
Staphylokinase
As another indirect plasminogen activator of bacterial origin, staphylokinase is secreted by lysogenic Staphylococcus aureus [53]. This protein generates plasmin only on the fibrin clot surface because of the rapid inhibition of free plasmin by its inhibitor α2-antiplasmin [54]. Staphylokinase also prefers conformation of the fibrin-bound plasminogen: only a small percentage associates with free molecules, but the main fraction strongly interacts with the fibrin-bound ones [55]. These features help prevent bleeding complications. Successful clinical application of staphylokinase for thrombolysis has been confirmed by the Collaborative Angiographic Patency Trial of Recombinant Staphylokinase (CAPTORS) I and II trials in patients with acute ST-elevation myocardial infarction [56,57].
Desmoteplase
Blood-sucking animals are a useful source of naturally evolved plasminogen activators. Desmoteplase, a 50 kDa serine protease, was discovered in the saliva of the vampire bat Desmodus rotundus in 1974 [58]. Desmoteplase consists of four functional domains: (1) a fibronectin-like or finger domain (F), (2) an EGF-like domain (E), (3) a kringle 1 domain (K1), and (4) a catalytic serine protease domain (P). The last one contains three catalytic AAs involved in the proteolysis: His237, Asp296, and Ser393 [59]. Desmoteplase adopts an open conformation upon binding to fibrin via the F domain, which increases its plasmin generating activity approximately 100,000-folds, the highest value among all thrombolytics discovered so far [60]. Substitution of the alteplase F-domain with the corresponding domain of desmoteplase and the removal of the K2 domain increased the affinity to fibrin by 1.2-fold and provided fibrin stimulatory effect of up to 1,560 times [61]. A similar F-domain replacement and deletion of the lysine binding site of the K2 domain resulted in a 1.2-fold greater affinity to fibrin and a 14-fold higher fibrin stimulatory effect [61,62]. Clinical trials, such as Desmoteplase in Acute Ischemic Stroke (DIAS) [63] and Dose Escalation of Desmoteplase in Acute Ischemic Stroke (DEDAS), have revealed that desmoteplase has high recanalization (up to 53.3% in patients treated with 125 µg/kg desmoteplase) and low intracranial hemorrhage (0%) rates [64]. DIAS-2 and DIAS-3 trials have confirmed that it is a safe thrombolytic drug [65].
Development of novel thrombolytics
Characteristics of “ideal” thrombolytics
Enzymatic thrombolysis has revolutionized the therapy of cardiovascular and cerebrovascular diseases; early treatment (1) restores blood flow in the occluded vessels, (2) reduces the damage of surrounding tissues, and thus (3) increases survival rates [46]. The “ideal” thrombolytic drugs should (1) provide fast reperfusion in all patients, (2) prevent reocclusions, (3) have high fibrin specificity for selective activation of clot-bound plasminogen to decrease bleeding complications, (4) be retained in the blood for a long time to minimize dosage and allow administration as a single bolus vs. continuous infusion, (5) be resistant to inhibitors, and (6) be non-antigenic for repetitive usage [66].
Shortcomings of current thrombolytics
Current FDA-approved thrombolytic drugs, such as streptokinase, urokinase, and alteplase, have several limitations, restricting the overall success rates of the treatments. Streptokinase has low fibrin specificity and causes an immune response in host organisms. Urokinase degrades not only fibrin clots but also fibrinogen and other plasma proteins, leading to systemic fibrinolysis and increased rates of bleeding complications [47]. Owing to low fibrin specificity, urokinase is not used for thrombolysis in stroke treatments. Instead, it is widely used for thrombolytic therapy of deep venous thrombosis, peripheral arterial occlusive disease, and pulmonary embolism [48,67,68], as described previously. It was also found to be effective for the restoration of patency in intravenous catheters occluded by clotted blood [69].
Alteplase has a short plasma half-life (approximately 6 minutes), but it was shown to be effectively (approximately 550-fold) stimulated by fibrin [70]. In fibrin-bound plasminogen, the target peptide bond becomes available because of conformational changes after lysine-dependent binding to fibrin [71]. These mechanisms make alteplase a safer thrombolytic drug in comparison with streptokinase and urokinase. Thus, it has a wider spectrum of FDA-approved clinical applications: it is used for the treatment of ischemic stroke, acute myocardial infarction, and pulmonary embolism [27,72]. However, in ischemic stroke trials the recanalization rate following alteplase administration was <10% within 1 hour, <35% within 2 hours, and <43% within 24 hours [73-75]. In approximately 3% to 6% of patients, alteplase treatments have led to intracerebral hemorrhage (ICH) [76,77]. The major predictive factors for alteplase failure were a bigger thrombus size, poor collaterals, a proximal location of thrombus, or prolonged time to treatment [78-80]. In patients with acute ischemic stroke due to the middle cerebral artery (MCA) occlusion, intravenous alteplase was shown to recanalize the artery when the length of thrombus did not exceed 8 mm [79]. The recanalization rate following alteplase treatment for thrombi with a distal location, e.g., distal M1 MCA was approximately 4.5 times higher than for those located at the internal carotid artery (46.4% and 10.9%, respectively) [80].
Methods for developing novel thrombolytics
Many strategies are used to develop novel drugs that lack the aforementioned shortcomings of currently used thrombolytic agents (Figure 2). The well-traveled path of finding molecules with thrombolytic activity in different organisms was followed by molecular biology methods to facilitate their discovery, such as construction of fusion proteins or substitution mutagenesis. Computational methods have been widely used in recent years.
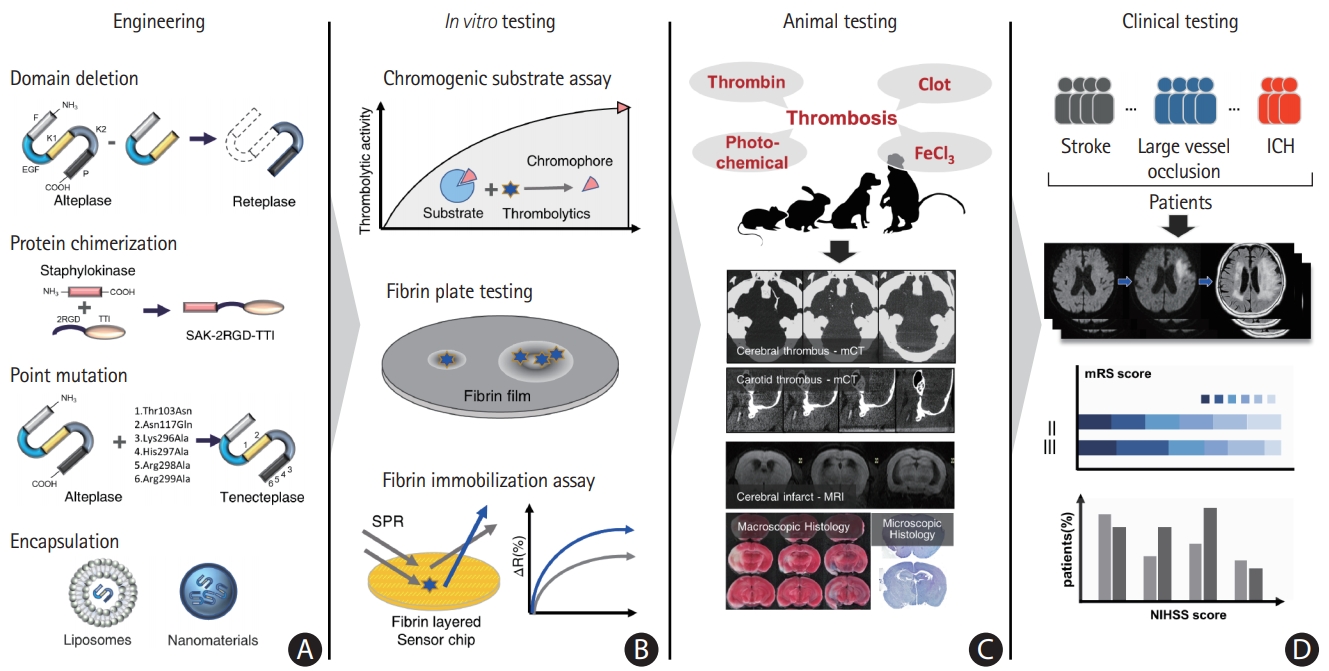
Scheme depicting the development of novel thrombolytics. The overall workflow is separated into four main steps, which are thoroughly discussed in the main text. (A) Development of novel molecules using protein engineering. (B) In vitro testing by biochemical assays. (C) In vivo testing in animal models. (D) Several phases of clinical testing. F, finger domain; EGF, epidermal growth factor-like domain; K1, kringle 1 domain; K2, kringle 2 domain; P, protease domain; RGD, arginine (R), glycine (G), aspartic acid (D); TTI, tsetse thrombin inhibitor; SAK, staphylokinase; SPR, surface plasmon resonance; mCT, micro computed tomography; MRI, magnetic resonance imaging; ICH, intracerebral hemorrhage; mRS, modified Rankin Scale; NIHSS, National Institutes of Health Stroke Scale.
Discovery of new molecules
Many enzymes and small molecules found in nature have thrombolytic activity. Several thrombolytics have been developed by the identification and characterization of molecules found in bacteria, such as the fibrin-specific thrombolytics from Streptococcus uberis [81], Bacillus [82], or Stenotrophomonas [83]. A significant effort is invested into nattokinase—a thrombolytic agent originating from Bacillus subtilis found in the fermented soybean food natto [84]. Other sources of thrombolytics are various eukaryotes, such as the Chlorella alga [85], venomous snakes [86,87], and polychaetes [88]. Thrombolytics of non-mammal origin are often direct fibrinolytics that cleave fibrin directly instead of activating plasminogen. The discovery of new thrombolytics can greatly benefit from next-generation sequencing technologies and gene-mining software tools [89]. Small molecules such as triprenyl phenolic compounds from the Stachybotrys mold promote the binding of plasminogen to fibrin and induce a conformational change in plasminogen, which promotes its activation and thus fibrinolysis [90].
Domain deletion
As mentioned before, alteplase is a protein with five exons coding for the domains of different functions [91]. Besides fibrinolysis, these domains are associated with neurological side effects such as blood-brain barrier dysfunction, N-methyl-D-aspartate receptor (NMDAR)-mediated neurotoxicity, and other neurological functions [92-94]. The first three domains interact with the mannose receptor and low-density lipoprotein receptor-related protein 1, which are related to tPA clearance by endothelial cells and hepatocytes, respectively [33,95,96]. Reteplase, a deletion mutant of alteplase, lacks the finger, EGF-like, and kringle 1 domains. It has a long half-life of 15 minutes owing to the absence ofthe domains responsible for receptor-mediated clearance. Compared with alteplase, reteplase has lower fibrin-binding and better clot penetrability, which leads to faster and more uniform thrombolysis and fewer reocclusions. Owing to the lack of glycosylation and fewer disulfide bridges, reteplase can be expressed at a lower cost using a prokaryotic system [97].
Protein chimerization
In contrast to avoiding unwanted functions by the aforementioned domain deletions, constructing chimeric proteins by fusing different proteins or their domains can couple more functions within a single protein. Fibrin-specificity can be achieved through the fusion of EGF domains of thrombomodulin or the K2 domain of alteplase on streptokinase [98,99]. A drug such as staphylokinase can become both thrombolytic and anticoagulative through the fusion of the arginine-glycine-aspartate tripeptide motif, hirudin, annexin XI, or the tsetse thrombin inhibitor [100-103]. Fusion of the fibrin-binding kringle 1 domain of plasminogen on staphylokinase increased fibrinolytic effectivity [104]. Fusion of the Gly-His-Arg-Pro (GHRP) peptide that has a binding affinity for fibrinogen enhanced the fibrin affinity of alteplase [105,106]. In addition, greater fibrin specificity was achieved by substituting the fibrin-binding fibronectin-like domain of alteplase with the analogous domain of desmoteplase [61]. Another interesting approach is the fusion of microplasmin and the activation-specific anti-glycoprotein IIb/IIIa antibody. In this way, fibrinolysis by microplasmin is targeted to the clot. Microplasmin is far less inhibited by α2‐antiplasmin than plasmin in the blood [107].
Point mutations
The very short half-life of streptokinase can be extended up to 21-fold by substituting specific charged residues responsible for its proteolysis or by PEGylation (the process of attaching polyethylene glycol [PEG] polymer to molecules) [108-110]. Staphylokinase can be induced to have reduced immunogenicity (30%) or improved fibrinolytic effectivity by substituting only a few specific residues and using PEGylation [111,112]. Alteplase can be made resistant to PAI-1 by mutating four charged residues in the catalytic domain to alanine. This mutation, combined with two more substitutions to alleviate low-density lipoprotein-like receptor 1-mediated clearance, provides TNK, which has a 4-fold increased half-life, with more resistance to PAI-1 and higher fibrin specificity compared with alteplase. These properties make TNK a promising new thrombolytic drug for treating ischemic stroke [113]. Single point mutations have been introduced in tPA to attenuate its NMDAR-mediated neurotoxicity [114]. Others showed that alteplase can be made 28-fold more fibrin specific by substituting the plasmin-sensitive cleavage site with that of desmoteplase [60]. Recently, Goulay et al. [115] engineered a variant of tPA devoid of its interaction with the NMDAR by substituting tryptophan 253 in the kringle 2 domain with arginine. They also introduced another point mutation to reduce tPA sensitivity to plasmin without affecting its fibrinolytic activity: a substitution of arginine 275 with serine. In a rat model of ICH and catheter-delivered in situ thrombolysis for the drainage of ICH, the variant tPA reduced hematoma volume. Furthermore, unlike alteplase, the mutant tPA also reduced peri-hematomal neuronal death and edema progression, probably due to the lack of promotion of NMDA-dependent neurotoxicity [115]. Urokinase became fibrin-specific with fewer bleeding complications by introducing point mutations [116].
Liposomes and encapsulation
Encapsulation of thrombolytics in liposomes or other nanomaterials offers interesting opportunities for improving thrombolytic therapy. Liposomes are spherical particles delimited by a lipid bilayer, with a size ranging from 30 nm to several micrometers. Inside the bilayer-delimited sphere, there is an aqueous phase in which therapeutic molecules, including thrombolytics, can be encapsulated. Other encapsulation techniques are used: encapsulation in blood cell-derived membranes or polymer-based nanoparticles.
Encapsulation in liposomes can significantly improve the half-life of thrombolytics by preventing interactions with clearance receptors in the bloodstream. It also limits systemic plasminemia by preventing the interaction of encapsulated thrombolytics with plasminogen not located in the thrombus [117]. The half-life and binding properties can be gradually tuned by engineering the composition of liposomes by altering the ratio of different phospholipids in their bilayer, coating them with ligands, or performing PEGylation [118].
The half-life of liposomal streptokinase is 16-fold longer [117]. The half-life of tPA was prolonged 21-fold by encapsulation in liposomes [119]. Besides increasing the half-life, liposomes can also provide other useful pharmacokinetic properties. The accumulation of PEGylated liposomes occurred in two different therapeutic windows of 0.5 and 48 hours after stroke [120]. The accumulation of liposomes occurred through transcellular transport during the first window and trans- and para-cellular pathways during the second window [120]. This could be used to provide targeted therapeutic action both spatially and temporally.
Liposomes themselves prevent immunogenicity and have anti-inflammatory properties. These properties are even more pronounced when they are loaded with anti-inflammatory or neuroprotective agents [121,122]. Encapsulation of dexamethasone or a combination of tPA and cyclosporine A in liposomes improved neurological scores and reduce subsequent neuroinflammation in rat models of ischemic stroke.
One of the most sought-after properties of liposomes and other nanoformulations is their ability to target thrombolytics to the clot. Liposomes decorated with a fibrinogen-mimicking peptide and loaded with urokinase bind to platelets with high specificity [123]. Urokinase encapsulated in liposomes decorated with this peptide produced a thrombolytic effect in a rat mesenteric thrombosis model with only 25% of the dose of free urokinase, and showed significantly less prolonged bleeding times than free urokinase [124]. Targeted liposomes loaded with urokinase showed improved thrombolytic efficacy in a rat embolic stroke model, too [124].
Thrombolytic action in the clot can also be achieved through targeted release using ultrasound, infrared radiation, temperature, or shear stress-activated liposomes and other nanotherapeutics [117,125-127]. The use of echogenic liposomes loaded with thrombolytics couples the diagnostic and therapeutic procedures and was shown to enhance thrombolytic potency [117]. Loading thrombolytics into blood cell membrane-based particles showed improved therapeutic efficacy over free thrombolytics in animal ischemic stroke and thrombosis models, while preserving coagulation profiles and having fewer bleeding complications [128,129]. Blood cell membrane-based nanoparticles coated with tPA have led to a significantly better neurological score and survival rate, with a practically unchanged coagulation profiles. Although many studies are exploring the encapsulation of thrombolytics in stroke treatment [117,118], the translation to clinical practice is still pending.
Rational protein design
Determination of the crystal structures of thrombolytic enzymes and using them for in silico modeling have led to big leaps in the development of thrombolytic agents [36]. The knowledge of the structure of staphylokinase has led to the identification of sites for PEGylation that could improve the half-life while preserving the effectivity of staphylokinase. The effect of mutations on streptokinase can be studied and predicted using the crystal structure of streptokinase bound to microplasmin [130]. The complete structure of alteplase has not been fully elucidated. However, in silico predictions using the low-resolution structures of alteplase could estimate how deleting any domain decreases its catalytic activity or how it is influenced by glycosylation or enhanced by fibrin [35,131]. Establishing the structure of reteplase by homology modeling based on alteplase led to improvement in the solubility and fibrin affinity of reteplase [132].
Each candidate thrombolytic agent should be characterized in vitro and in vivo and compared with previous ones. Choosing efficient and effective experimental methods is important for characterization. The choice often poses a tradeoff between the efficiency and thoroughness of testing. A good in vitro testing strategy can screen more molecules and provide a higher chance of finding a good hit, while also testing candidate thrombolytics for many of the biophysical, biochemical, and biological aspects that are required to be a successful thrombolytic agent.
In vitro testing of novel thrombolytics
In vitro testing of thrombolytics is typically based on the physiological fibrinolytic pathway (Figure 3A); however, individual assays differ in the level of their complexity, analysis time, reproducibility, and the targeted property [133,134]. Since none of these methods is superior, it is necessary to always choose the most relevant ones for a given purpose. To obtain a complex understanding of the studied thrombolytics, a combination of different methods may need to be used. An overview of the available assays is provided in Table 1.
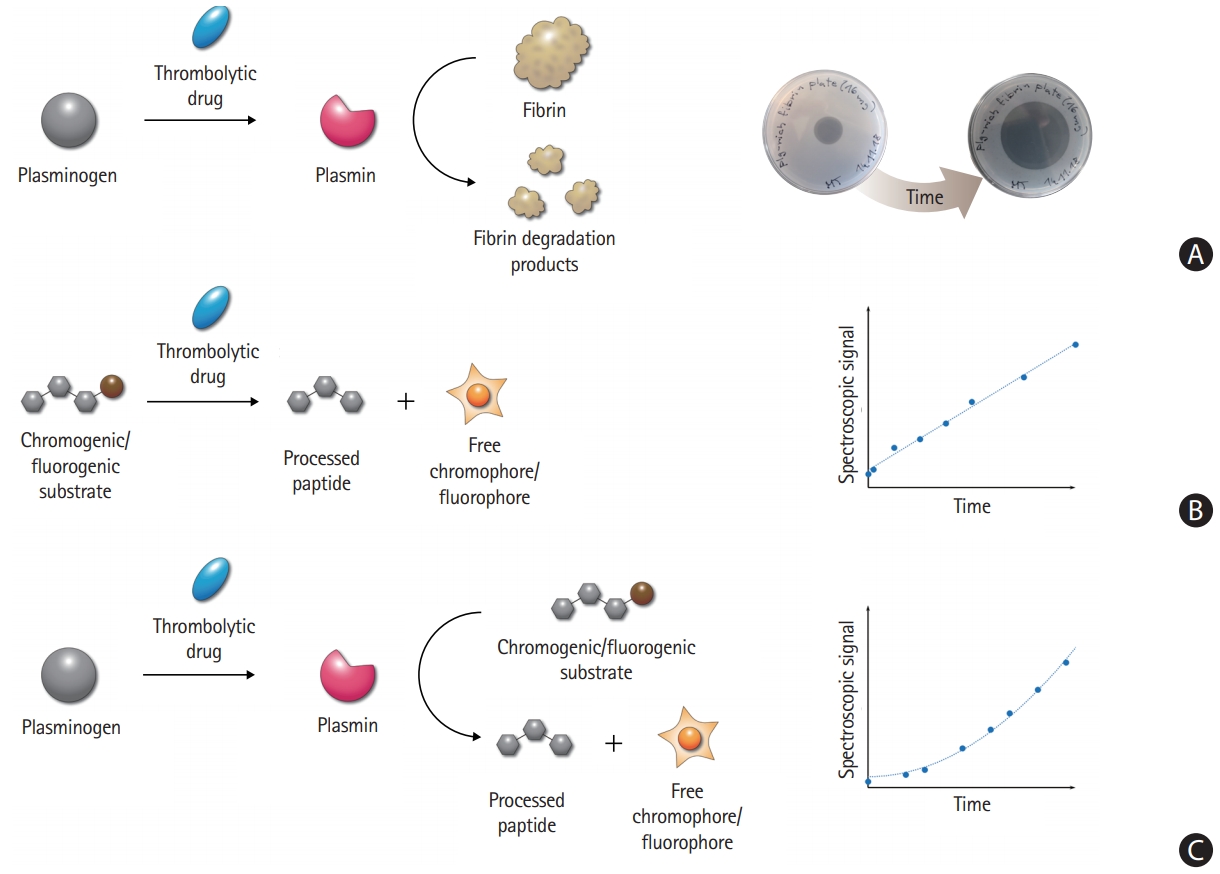
Overview of thrombolytic enzymatic activity assays. (A) The physiological thrombolytic pathway without any modification can be monitored by fibrin plates, providing clear zones through the dissolution of fibrin. (B) Direct chromogenic/fluorogenic assay uses a synthetic substrate instead of plasminogen. The assay allows easy and straightforward analysis of thrombolytic enzyme kinetics by a spectrophotometer. The direct cleavage by plasminogen activators results in a linear increase of the signal over time. (C) Coupled chromogenic/fluorogenic assay uses a synthetic substrate instead of fibrin, while the analyzed step of activation involves the physiological plasminogen substrate. The measurement is thus more robust, reliable, and closer to real thrombolytic events than the direct assay. The coupled nature of the assay results in a quadratic increase of the signal over time.
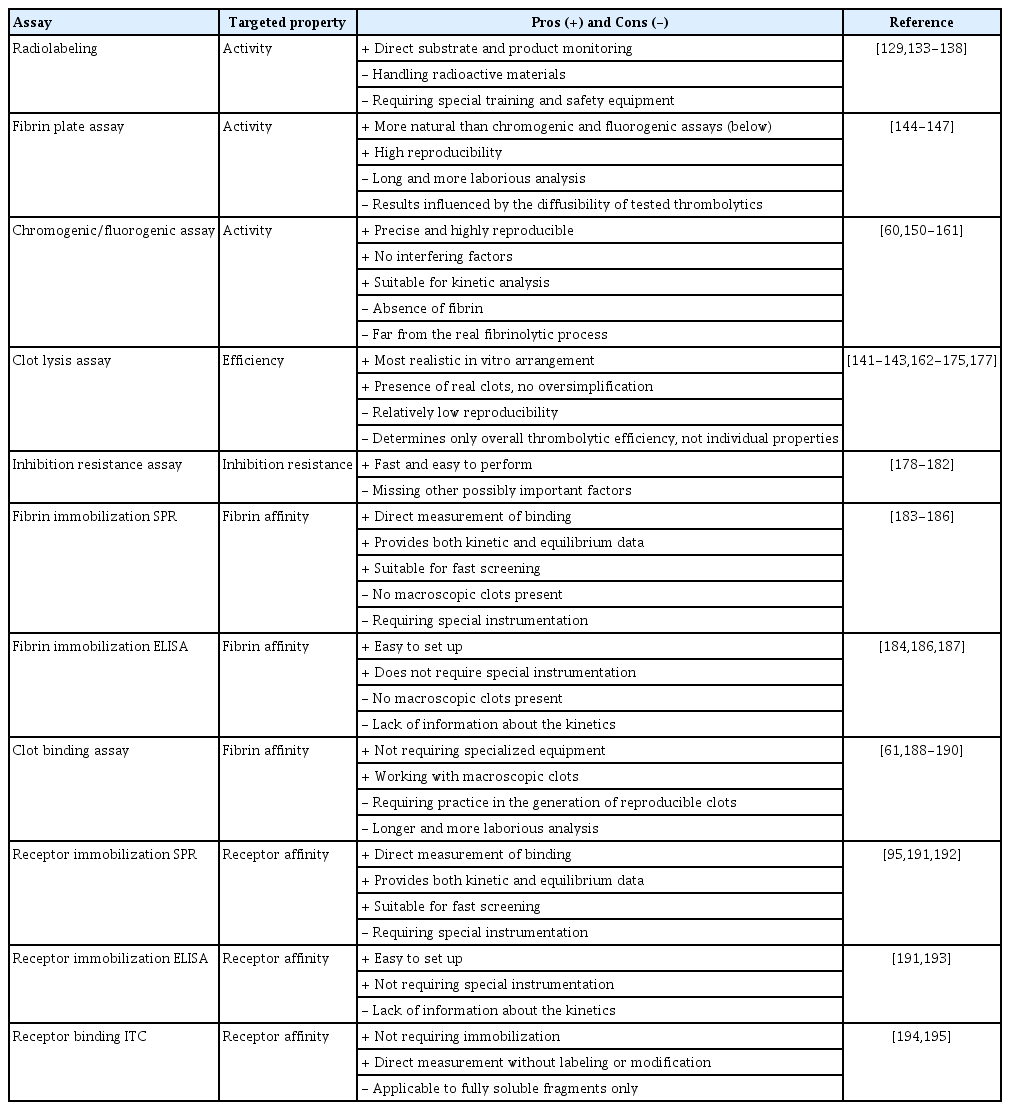
Comparison of the most common assays used for in vitro testing of thrombolytics
Enzymatic activity assays
Numerous methods for the assessment of the fibrinolytic potential of thrombolytic proteins are available, ranging from the historic radiolabeling [70,135-140] to more advanced methods, such as the euglobulin lysis assay, clot formation and lysis (CloFAL) assay, and streptokinase-activated lysis time (SALT) assay [141-143]. Three basic principles are applied in most of the developed assays.
Fibrin plate assay
The fibrin plate assay relies on the physiological fibrinolytic pathway (Figure 3A). The plates are prepared by mixing melted agarose with plasminogen, thrombin, and fibrinogen. When a thrombolytic agent is added, fibrin dissolution is assessed by measuring the extent of the clear zone [144-147]. This method uses fibrin as a key component, making the assay more natural than the chromogenic/fluorogenic assay (see below), while still providing robust and reproducible results. In contrast, the fibrin plate assay is more laborious and time-consuming. Moreover, the growth of the zones depends not only on the fibrinolytic activity but also on the diffusibility of the tested thrombolytics. Modified methodologies, such as the dyed fibrin plate assay [148] and fibrin microplate assay [149], have been developed to resolve some of these problems.
Chromogenic/fluorogenic substrate assay
This method is based on the cleavage of a synthetic chromogenic or fluorogenic peptide substrate by a thrombolytic enzyme (Figure 3B and C), releasing a free chromophore or a fluorophore and thus generating a measurable spectroscopic signal [150-157]. The assay is fast, highly reproducible, and depends solely on the activity of the tested enzyme. However, the assay does not use fibrin, a key component influencing thrombolytic activity. To account for the stimulatory effect of fibrin, it is possible to add a small fibrin clot, fibrinogen, or fibrin fragments into the reaction mixture [60,158-161]. The synthetic substrate can be cleaved directly by plasminogen activators (Figure 3B) [152,155-157], or in a coupled arrangement. A thrombolytic drug first activates the natural substrate plasminogen to plasmin, which subsequently processes an analogical chromogenic/fluorogenic substrate (Figure 3C) [153,156,157]. The use of the coupled arrangement makes the analysis more reliable.
Clot lysis assay
In this assay, macroscopic clots are prepared either by mixing thrombin with plasminogen and fibrinogen [162,163], or clotting human plasma or blood [141-143,164-170]. Thrombolytics-induced lysis can be roughly monitored visually to report the complete clot lysis time [141,142,162-164] or more accurately, by the change in clot-related light scattering [141,143,169,171]. Other options include measuring (1) the release of red blood cells (RBCs) from whole blood clots [165,166,172,173], (2) changes in the clot mass [167,168], (3) the viscoelastic properties using thromboelastography [174,175], or (4) fluorescence change upon dissolution of labeled clots [176,177]. Unlike the chromogenic/fluorogenic substrate assay and the fibrin plate assay, these assays use macroscopic clots, thereby better mimicking in vivo biology. However, a higher level of procedural complexity decreases the accuracy and reproducibility of this assay. In addition, the assays do not solely monitor the enzymatic activity of thrombolytics, but rather their overall effectiveness.
Inhibition resistance assays
Inhibition resistance assays are performed by incubating thrombolytics with an inhibitor at a predefined concentration for a predefined time. Thereafter, the amount of free and active thrombolytics is determined using enzyme-linked immunosorbent assay (ELISA) [178,179] or the chromogenic/fluorogenic assay [178,180-182]. Such experiments provide information about both the kinetics and thermodynamics of inhibition.
Fibrin affinity assays
Fibrin immobilization assay
Fibrin layer or fibrin fragments can be immobilized on sensor chips, and the binding of thrombolytic drugs can be directly analyzed using surface plasmon resonance (SPR). This measurement provides information about both the affinity and the kinetics of the interaction [183-186]. Alternatively, fibrin can be immobilized on the surface of a microplate well, incubated with the tested thrombolytic agents, and the amount of bound molecules is determined by using ELISA or the chromogenic/fluorogenic assay [184,186,187].
Clot binding assay
To analyze the affinity to macroscopic fibrin clots, fibrinogen is mixed with the tested thrombolytic agents, and clotting is initiated by thrombin. The clot is subsequently sedimented using centrifugation, and the supernatant is used for measuring the amount of unbound thrombolytics using ELISA, the fibrin plate assay, or the chromogenic/fluorogenic assay [61,188-190]. Although this assay requires thorough optimization to obtain reproducible results, it uses macroscopic clots and does not require any complex instrumentation.
Receptor binding assays
The affinity of thrombolytics to membrane receptors is typically determined analogically as the affinity to fibrin layers. One option is to analyze binding affinities and kinetics using SPR [95,191,192]. An alternative approach is to measure the amount of thrombolytics bound to immobilized receptors using specific antibodies (ELISA arrangement) or by the chromogenic/fluorogenic assay after a washing step [191,193]. In addition, there have been studies reporting affinities to soluble receptor fragments, measured by isothermal titration calorimetry [194,195].
Overall, in vitro methods are useful for assessing the individual properties affected by structural modifications of a thrombolytic agent. In addition, the assays serve as an important platform for screening large libraries of designed drug variants in a cost-effective, time-efficient, and ethical manner. However, simplifications and deviations from physiological conditions make it essential to further test the safety and efficacy of candidate thrombolytics using in vivo animal models.
In vivo testing of novel thrombolytics
Thromboembolic stroke models, in which preformed blood clots are injected in arteries, have been generated most often in rodents (rats [196-212] and mice [177,213-219]) and other species including rabbits [220-224], dogs [225,226], zebrafish [227], and non-human primates [228,229]. In this section, we summarize the current available thromboembolic stroke models and their advantages and disadvantages in testing thrombolytic agents.
Clot injection-induced thromboembolic stroke model
Embolic stroke is induced by injecting preformed (autologous or heterologous) blood clots into the MCA—anterior cerebral artery bifurcation area [230-233]. Despite the relatively high mortality due to invasive surgical procedures, these animal models closely mimic human clots with high variability in the biochemical or cellular composition, and size [234].
The biochemical and cellular composition can be manipulated by adding fibrin or thrombin to the blood before clotting [205] or by mixing the blood with RBCs or platelets [235]. The diameter or length of clots can be controlled by using a clot-forming tube or catheter in various diameters, or by cutting clots in different lengths [205,212,236]. Strict control of the molecular and cellular properties of clots could reduce the experimental variability and increase the reproducibility in the investigation of thrombolytic agents. However, spontaneous recanalization or multifocal distal embolization after intra-arterial injection and the circle of Willis anatomy variations could have variable effects on the location and extent of the resulting cerebral infarction.
We previously showed that the heterogeneous nature of a mouse embolic stroke model could be characterized and managed by using a combined approach of in vivo cerebral blood flow (CBF) monitoring and ex vivo near-infrared fluorescent imaging to visualize thrombus [216,217]. More recently, we developed fibrin-targeted glycol-chitosan-coated gold nanoparticles (GCAuNPs) for computed tomography (CT)-based direct imaging of cerebral thromboembolism and carotid thrombosis [217]. In a follow-up study, thromboembolic burden and the efficacy of tPA therapy could be assessed serially, noninvasively, and quantitatively using microCT and the fibrin-targeted AuNPs in mice [237].
In situ thrombosis models
Thrombin-induced thromboembolic stroke model
Purified thrombin injection into the lumen of the MCA exposed by excising the dura after performing a small craniotomy leads to in situ thrombosis, followed by CBF reduction and cerebral infarction [230,232,238]. Thrombi that were produced by this method in Swiss mice consisted mainly of polymerized fibrin containing a low number of platelets [232]. This composition does not seem to accurately reflect that of clots in human ischemic stroke. Approximately 75% of the clots retrieved using the Merci device showed platelet/fibrin accumulation, linear neutrophil/monocyte deposition, and erythrocyte-rich accumulation [239]. However, another study demonstrated a highly complex ultrastructure of human clots with a diverse organization of fibrin, platelets, and RBCs within each thrombus and across various thrombi [240]. Injecting FeCl3 instead of thrombin could generate platelet-rich cerebral thrombi, providing an opportunity for studying the influence of different clot components on the efficacy of a thrombolytic agent [230].
Mortality rates are lower owing to the less invasive nature of the surgical procedure compared with the intra-arterial clot injection-induced thromboembolic stroke model. In addition, site-specific thrombin injection-mediated accurate and consistent clot localization may ensure more reproducible infarct volumes. However, a study characterizing this model in C57 black/6J mice (n=126) showed that 65% of animals (n=82) were excluded because of an unsuitable location of the MCA bifurcation for thrombin injection, bleeding complications, or spontaneous recanalization within 20 minutes (n=26). Only 44 animals demonstrated stable clot formation and cortical infarction. Spontaneous recanalization within 24 hours was observed in non-human primates as well as in mice [241], complicating the analysis and interpretation of the therapeutic efficacy of a thrombolytic agent. Moreover, this model suffers from the difficulty of assessing functional neurological deficits. However, a previous study demonstrated the utility of this model for comparing the efficacy of alteplase on infarct volume at different doses (0.9, 5, and 10 mg/kg) and at different times after stroke onset (from 20 minutes to 4 hours). At either high (5 to 10 mg/kg) or low (0.9 mg/kg) doses of alteplase, early (<3 hours) treatment significantly reduced the final infarct volume, whereas late administration showed no effect [242].
FeCl3-induced thromboembolic model
Thrombosis is induced by topical application of 2.5% to 35% ferric chloride (FeCl3) to the artery or vein after surgically exposing the target vessel. The generally accepted pathomechanism of FeCl3-induced thrombosis is oxidative vascular wall injury, causing endothelial denudation and in situ thrombosis. As another possible mechanism [243,244], micromolar levels of ferrous ions permeate the endothelium, and the ions in the bloodstream form agglomerates with platelets, RBCs, and plasma proteins, which subsequently induces thrombosis. Activated platelets, fibrin strands, and entrapped erythrocytes were found on the endothelial surface of the rat carotid artery 10 minutes after 35% FeCl3 application [245]. The clot size, degree of vessel occlusion, and blood flow reduction can be managed by adjusting the concentration and duration of the FeCl3 application. In a later dose-ranging study [246], 2.5% FeCl3 applied to the mouse carotid artery for 10 minutes was sufficient to occlude blood flow completely. Topical application of 10% FeCl3 to the distal MCA for 3 minutes induced thrombus formation, CBF reduction, and ischemia without hemispheric swelling, hemorrhage, or mortality by 24 hours. Because of rapid thrombus formation at a pre-defined location with high reproducibility in the clot size, this model has been widely used for cardiovascular research.
Previously, we developed the first hyperacute direct thrombus imaging technique using CT and thrombus-seeking GC-AuNPs, which enables in vivo assessment of thrombus burden/distribution and characterization of its evolution promptly and quantitatively [247]. MicroCT imaging could visualize both the presence and extent of thrombi in mouse carotid arteries without a single failure of detection. The AuNP thrombus imaging enabled the monitoring of thrombus evolution (either spontaneous or post-tPA), which could be mapped at high resolution in both space and time. A long circulating half-life (up to 3 weeks) of GC-AuNPs allowed for repetition or ongoing monitoring of thrombogenesis and thrombolysis without repeated injections of the imaging agent.
Photochemical thrombosis model
Intravenous injection of a photosensitive dye (e.g., Rose Bengal or erythrosine B) and light illumination to the cortical cerebral artery induces in situ thrombosis, CBF reduction, and cerebral infarction [248-258]. Light can be illuminated through the thinned skull of the animal brain without craniotomy. Dye-light interaction generates reactive oxygen species that causes endothelial damage and platelet aggregation, leading to the formation of a platelet-rich thrombus at the site of the photochemical reaction.
A recent study using this model [252] showed that infarct size was not affected by depletion or functional inhibition of platelets. The authors suggested potential mechanisms, such as blood-brain barrier disruption-mediated parenchymal edema causing secondary vascular compression as well as inflammatory brain damage involving cytotoxic radicals, microglial activation, and cytokine release [259-261].
The major strengths of the photothrombosis model include reproducible infarct size and location as well as a relatively simple surgical procedure and low operative mortality. The infarct size can be controlled by changing the concentration of Rose Bengal and the duration of light illumination [258,262-269].
Poor responses to tPA-mediated thrombolytic therapy, which is likely due to a low fibrin content in the platelet-rich clots, was improved by increasing the fibrin content by mixing Rose Bengal (50 mg/kg) with thrombin (80 U/kg) [270].
Lu et al. [255] showed that photochemical thrombotic stroke could be induced in conscious animals. They mounted a wearable miniature headstage, which allows for both light illumination (532 nm) to the MCA and laser speckle contrast imaging [271] on the head of rats. This model better reflects most of the clinical stroke, which does not occur in anesthetic conditions, providing a unique research platform to study stroke pathophysiology without the confounding effects of anesthesia.
Clinical testing of novel thrombolytics
Alteplase significantly improves post-stroke functional outcomes; however, potential risks, particularly symptomatic intracranial hemorrhage, can complicate intravenous tPA therapy [2,77,272]. As described before, TNK compared with alteplase has a higher affinity for fibrin and less impairment of hemostasis. TNK has greater resistance to PAI-1 and a longer plasma halflife, allowing single intravenous bolus injection [273]. Desmoteplase is another new thrombolytic agent with a longer half-life and higher fibrin specificity than alteplase [65].
In the Australian TNK trial, TNK 0.1 and 0.25 mg/kg, compared with alteplase, were associated with better clinical outcomes in 75 patients with large artery occlusion, probably due to higher reperfusion rates and more favorable perfusion patterns within 6 hours from symptom onset [274]. However, the Tenecteplase versus Alteplase for Management of Acute Ischemic Stroke (NORTEST) study investigators reported no difference between TNK 0.4 mg/kg and alteplase in terms of the safety and efficacy in 1,100 patients who were eligible for IV thrombolysis within 4.5 hours of onset [275]. The majority of participants had a mild stroke (median National Institutes of Health Stroke Scale of 4), which complicates the interpretation of the results.
With the advent of endovascular thrombectomy, Extending the Time for Thrombolysis in Emergency Neurological Deficits–Intra-Arterial (EXTEND-IA) TNK trial compared TNK 0.25 mg/kg with alteplase in patients with large vessel occlusion who were scheduled for thrombectomy. TNK was superior to alteplase in the recanalization on the initial angiogram (22% vs. 10%). In addition, TNK achieved a more favorable 90-day functional outcome in the ordinal analysis of the modified Rankin Scale score (common odds ratio, 1.7; P=0.037) [73].
Clinical trials of desmoteplase selected patients who came in late time windows but had salvageable tissue on brain imaging. In the Study of DIAS-2 trial, 90 and 125 μg/kg of desmoteplase did not increase the clinical response rates at 90 days compared with the placebo in patients with ischemic stroke within 3 to 9 hours after symptom onset [276]. The DIAS-3 trials involved 492 patients with high-grade stenosis or occlusion of major cerebral arteries within 3 to 9 hours after symptom onset. Desmoteplase (90 μg/kg) and the placebo did not differ significantly in terms of the primary outcome: the proportion of patients with a favorable modified Rankin Scale score (0–2) at 90 days (51% vs. 50%, respectively) [65]. The DIAS-4 trial was terminated early, and the combined results of DIAS-3, DIAS-4, and DIAS-J (Japan) were published [277]. Late treatment with intravenous 90 μg/kg desmoteplase was safe with an increased rate of recanalization, although it did not significantly improve the functional outcome at 3 months [277]. The limitations of the studies were: (1) focusing on the late time window, (2) without consistent application of imaging criteria for patient selection, and (3) sample size-related issues in DIAS-2 (small sample size) and DIAS-3 (21% patients excluded from the final analysis).
To date, TNK and desmoteplase are the only drugs that have undergone phase II clinical trials. In terms of safety and efficacy, clinical trials did not definitively prove the superiority of TNK over alteplase [73,272-275,278,279]. A meta-analysis demonstrated a similar rate of symptomatic intracerebral hemorrhage (sICH) between TNK (3%) and alteplase (3%) [280]. Although two trials (Australian TNK and EXTEND-IA TNK) have proven the superior efficacy of TNK over alteplase, the meta-analysis did not show the overall superiority of TNK. Ongoing large sample-sized trials (Alteplase-Tenecteplase Trial Evaluation for Stroke Thrombolysis [NCT02814409] and Tenecteplase versus Alteplase for Stroke Thrombolysis Evaluation TASTE-2 [ACTRN12613000243718]) comparing the safety and efficacy of TNK versus alteplase may provide more conclusive data [273]. Previous studies suggest that desmoteplase and alteplase might have comparable safety and efficacy, although a head-to-head study has not been conducted.
As described above, in the Australian TNK and EXTEND-IA TNK trials, TNK achieved a higher recanalization rate compared with alteplase [274]. In TNK-tPA Evaluation for Minor Ischemic Stroke With Proven Occlusion (TEMPO-1) trial, TNK demonstrated a remarkable recanalization rate in patients with mild stroke and intracranial arterial occlusion [273]. Desmoteplase also showed an outstanding recanalization rate in the DIAS-3 trials [65]. Thus, it is likely that patients with large vessel occlusion due to large or long thrombi may benefit more from recanalization therapy using the new thrombolytics. An ongoing TEMPO-2 trial (NCT02398656) may contribute to this issue.
Although the era of endovascular therapy has finally come, intra-arterial mechanical thrombectomy cannot and will not be performed in all patients with acute ischemic stroke. Intravenous thrombolysis may facilitate subsequent mechanical thrombectomy and can be performed promptly while preparing the intra-arterial intervention. Moreover, the chemical lysis of microthrombi as well as the mechanical recanalization of large arteries is required for optimal cerebral reperfusion, which is crucial for improving clinical outcomes. In addition, in the setting of the drip-and-ship approach, a single bolus injection (of a new thrombolytic drug) will have a clear logistical advantage over the continuous infusion (of tPA).
Conclusions and perspectives in the development of thrombolytics
Despite a large variety of biochemical features of natural and engineered thrombolytic enzymes, none have demonstrated “optimal” clinical outcomes. A missing piece of information that will be useful for the development of more efficient thrombolytics is the complete tertiary structures of alteplase, TNK, and desmoteplase. These structures have not been determined so far, and computer models do not provide the necessary molecular details. Particularly, it would be informative to know the structures of their complexes with fibrin and/or plasminogen, which would allow for more effective engineering of the affinity and specificity. We are witnessing improvements in structural techniques, such as cryo-electron microscopy, X-ray crystallography, and nuclear magnetic resonance (NMR) spectroscopy, which can reveal the structures of even large multi-domain and multi-protein complexes.
We see a duality in the current trends in the development of novel thrombolytics. On the one hand, novel thrombolytics are being developed using more precise design strategies. Research and developmental strategies in this field have moved from deleting and fusing whole domains of proteins to introducing point mutations. Researchers are increasingly using the methods of rational protein design, which are more specific, focused, and reliable. On the other hand, the vastly expanding repertoire of deoxyribonucleic acid sequences and small molecules enables the discovery of novel thrombolytics in biological samples collected from living ecosystems. We envision that future thrombolytic formulations can be developed by (1) computational rational design that is based on new structures identified by protein crystallography, NMR spectroscopy, and cryo-electron microscopy; (2) discovering novel thrombolytics using databases of protein sequences and small molecules; (3) combining with antioxidants or neuroprotective drugs that could potentially attenuate reperfusion injury, and (4) improving the delivery of thrombolytics via liposomes or other nanocarriers.
One of the attractive strategies for improving thrombolytic treatment is the use of sonothrombolysis [281]. To test newly designed thrombolytics under these specific conditions, progressive and reliable in vitro methodologies combining standard assays and ultrasound devices are being developed [282-285]. At the same time, the development of advanced in vitro microfluidic models simulating real arterial events are of large interest [286-292]. Thanks to peristaltic pumps circulating blood inside microfluidic chips with blood vessel-like channels coated with endothelial cells, the in vitro testing is becoming highly reliable and relevant to clinical practice [286,289-291]. Once it is confirmed that these assays provide robust results, this approach might represent an effective and ethical alternative to animal testing. Moreover, substituting animal blood with human blood in the microfluidic system could lead to even more relevant results. The development and use of such advanced methods are crucial for the production and robust screening of newly discovered or rationally engineered thrombolytic proteins.
Clinical research has been based mainly on the use of a single thrombolytic agent, but precisely defined cocktails of multiple drugs might have better joint effects for thrombolytic therapy. The most effective combinations should be explored in vitro and in vivo using realistic animal models [293]. Finally, we would like to underline the critical importance of clinical trials; their unbiased performance is crucial for effective decision-making.
Notes
Disclosure
The authors have no financial conflicts of interest.
Acknowledgements
The authors would like to express their thanks to the Czech Ministry of Education for financial support (CZ.02.1.01/0.0/0.0/16_026/0008451). Dmitri Nikitin is the recipient of an individual grant MSCA fellowship (CZ.02.2.69/0.0/0.0/19_074/0016274), and Martin Toul is supported by the scholarship Brno Ph.D. Talent. Dong-Eog Kim is supported by grants from the Global Research Laboratory program (NRF-2015K1A1A2028228) and Basic Science Research program (NRF-2020R1A2C3008295), and Seungbum Choi from the Basic Science Research program (NRF-2019R1C1C1002909), funded by the Korean government. The authors also thank Su-Kyoung Lee for improving the figures and Dr. Jung E. Park for critical revision of this paper.