Introduction
In recent decades, experimental and clinical studies have demonstrated that there is an “invisible” cross-talk between the heart and the brain [1]. Even in the absence of cardiac disease [2], acute brain injury (ABI) can induce cardiac dysfunction, which in turn can increase mortality and lead to potentially long-lasting cardiac complications, such as heart failure [3]. Indeed, ABI such as ischemic stroke, seizures [4], aneurysmal subarachnoid hemorrhage (SAH) [5], and traumatic brain injury (TBI) [6] may result in challenging heart pathologies, such as neurogenic stress cardiomyopathy (NSC) [4,7,8].
In addition to the axis from the brain to the heart in patients with acute neurological injury, “heart to brain” connections must be seriously considered [9], especially in patients with cardiac dysfunction secondary to acute brain damage. Unfortunately, the pathophysiology of these heart-brain interactions is still poorly understood [1] and various factors have been suggested to explain the observed “heart to brain” cross-talk, including hemodynamic disturbances and impaired cerebral perfusion pressure (CPP) with subsequent cerebral hypoperfusion, development of secondary cerebral cardioembolic complications, oxygenation disturbances, neurohormonal mechanisms, systemic and cerebral inflammation [10], disruption of the blood-brain barrier (BBB), and activation of glial cells [2,11-15].
The present review contains a detailed discussion of heart-brain connection in patients with NSC due to isolated ABI with focus on hemorrhagic and ischemic stroke. In addition, the review highlights underlying pathophysiological mechanisms regarding the stroke-heart-brain axis leading to secondary and delayed cerebral injury and identifies further areas of research that could potentially improve outcomes in this specific patient population.
NSC: a complex definition
Cardiac dysfunction from neurological injury has been classified as neurogenic stunned myocardium (NSM) [7], which refers to a neurocardiogenic interaction in patients with ABI and results in reversible myocardial dysfunction [7,16] or NSC (i.e., takotsubo cardiomyopathy [TTC], secondary to neurological disorders) [17-19]. However, it is still unclear whether NSM and NSC are distinct pathological entities or divergent manifestations of the same pathophysiological process [1]. In addition, the terminology of myocardial dysfunction in patients with acute neurological injury is confusing and is still the subject of scientific debate [19]. Furthermore, it has been suggested that, since the terminology “stunned myocardium” generally refers to reversible myocardial dysfunction in the context of coronary artery occlusion with subsequent reperfusion, it would be preferable to use the term “stressed myocardium” in order to emphasize the pathophysiological causal association of the catecholamine storm phenomenon observed in these patients [5].
Moreover, the term “neurogenic stunned myocardium” comes from a time when clinicians were unfamiliar with the different types of TTC. As the definition of NSM is in accordance with the diagnostic criteria of TTC, a recent review [19] has recommended the use of the term “takotsubo cardiomyopathy secondary to neurological disorders,” together with a reference to its exact type, that is apical, basal, midventricular, or focal [19].
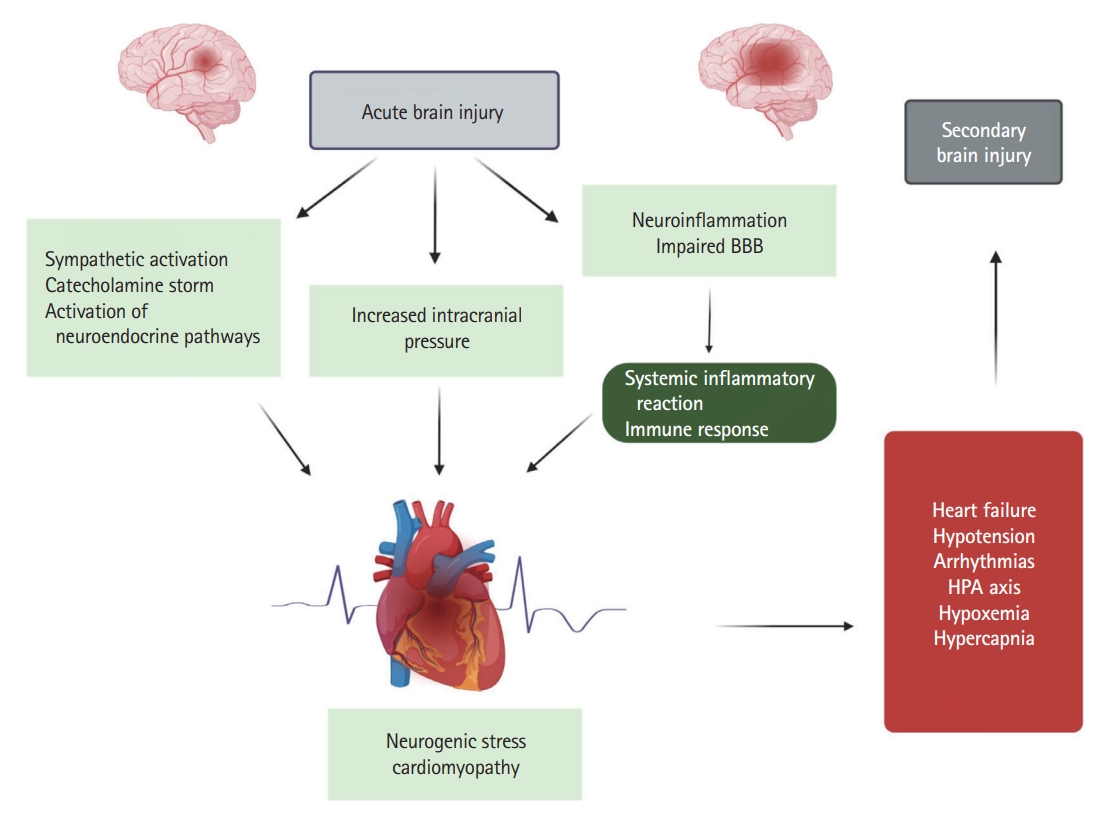
The pathophysiology of neurogenic cardiac injury in patients with and without pre-existing heart diseases is complex [18,19] and includes the principle pathophysiological mechanisms, such as autonomic dysregulation, excessive catecholamine release, myocyte injury, mitochondrial dysfunction, and perpetuated inflammatory cascades [9]. Given the etiological association of the development of TTC in patients with acute neurological or psychiatric disease (Figure 1) [20] and in order to emphasize the underlying pathophysiological mechanisms, the authors of the present study propose the use of the term “neurogenic stress cardiomyopathy” as a specific variant of TTC in acutely affected neurological patients.
NSC in patients with stroke
Pathophysiology of cardiac dysfunction in stroke
As mentioned above, the pathophysiology of neurogenic cardiac injury is complex [19] and, in addition to the above-mentioned pathophysiological mechanisms, includes stroke-induced disturbances of hypothalamic-pituitary-adrenal (HPA) axis and perpetuated local and systemic inflammatory cascades [9,18,19,21]. The consequences are increased myocardial demand, coronary microvascular spasm, acute and chronic structural and functional abnormalities, and molecular signaling myocardial alterations including microvascular endothelial disturbances, altered calcium metabolism, myocyte injury, contraction band necrosis, myocardial inflammation and fibrosis, oxidative stress, altered mitochondrial function, apoptosis and autophagy, and impaired gene expression [19,21-23].
Regarding NSC, despite extensive research, many questions remain poorly understood. However, the most prevailing theory trying to explain its pathophysiology is a sympathetic hyperstimulation due to physical and emotional triggers with an excessive release of catecholamines leading to direct catecholamine-toxicity, epicardial and microvascular coronary vasoconstriction, and adrenoreceptor-mediated damage [22,24]. Indeed, experimental models of SAH suggest an abrupt elevation of catecholamines within minutes following stroke, which positively correlates with the peak values of troponin T and creatine kinase-MB [25], phenomenon that may explain the increased frequency and severity of cardiovascular complications in the first three days after a stroke onset [26-28]. The typical distribution of left ventricular (LV) β-adrenergic receptors with apical-basal gradients and sympathetic innervation might explain the LV wall motion abnormalities seen in NSC as the apex characterized by increased density of receptors [29]. Indeed, the specific topographic distribution of β-receptors seems to be largely causally related to the increased apical responsiveness observed in states of catecholamine excess and especially epinephrine [2]. At the cellular level, the local action of catecholamines is facilitated by the close proximity of cardiomyocytes and nerve endings leading to impaired calcium homeostasis and altered β-adrenergic signal transduction, which in turn lead to myocardial contraction band necrosis and impaired coronary microcirculation [22,30,31]. Moreover, supra-physiological levels of epinephrine may have a negative inotropic effect, as they contribute to the switching of β2-adrenergic receptors from canonical stimulatory G-protein-activated cardiostimulant to inhibitory G-protein-activated cardiodepressant pathways [32]. The major pathophysiological aspect of altered function within the brain-heart axis in the pathogenesis of stroke-associated NSC is further supported by experimental and clinical research evidence which indicates a correlation between the development of cardiac dysfunction and ischemic events involving the insular cortex, especially the right insular cortex [33-35]. Indeed, current neuroimaging studies using functional magnetic resonance and positron emission tomography have confirmed the insular cortex as a cardinal regulator of the central autonomic nervous system [36]. The latter observation is crucial if taken into account that blood supply of insular cortex comes from the middle cerebral artery making it particularly susceptible to cerebrovascular disease [37].
Finally, systemic inflammation and more specifically the excessive release of interleukin (IL)-1 along with neurohormonal mechanisms seem to play a cardinal role in the development of NSC. Similarly to catecholamines, cytokine release occurs shortly after stroke; despite that elevation of their plasmatic levels lasts only few hours to days cytokine-induced myocardial remodeling can be preserved for months [19,22,23]. Further, parasympathetic dysfunction observed in patients with severe neurological damage such as SAH, intracerebral hemorrhage, and acute ischemic stroke may lead to myocardial damage through uncontrolled release of inflammatory mediators [38]. Additionally, recent research suggests that microglia, the brain-resident immune cells, play a pivotal role in regulating sympathetic nervous system activity (i.e., one of the main contributors of pathological cardiac remodeling) by release of a variety of substances, including chemokines, cytokines, and growth factors [39]. Moreover, massive release of epinephrine and norepinephrine has also a neurohormonal basis through the activation of the HPA axis and release of adrenocorticotropic hormone, sympathetic stimulation of the adrenal glands, elevation of plasmatic cytokines affecting the HPA axis leading to peripheral arterial vasoconstriction, sudden LV afterload increase, and elevation of LV end-systolic pressure [40-43]. Future research regarding the development of new therapeutic targets, such as blockade of β-adrenergic receptors or IL-1-inhibition combined with the precise elucidation of the exact pathophysiological pathways, may be crucial in preventing and managing cardiac damage in patients with stroke.
Clinical presentations of NSC in patients with stroke
It is generally accepted that stroke patients develop a two-way interaction between brain and heart, which makes them highly susceptible to severe cardiovascular complications [44]. Although emotional distress [45] and SAH [46] are the major causes of NSC, cardiac dysfunction may also occur after various neurological pathologies, such as TBI, hemorrhagic and ischemic stroke, central nervous system [47] infections, and epilepsy [48]. In general, cardiac manifestations in patients with stroke and NSC are highly variable and are associated with electrocardiographic (ECG) signs, such as tachyarrhythmias, QT-interval prolongation, T-wave inversion, and ST-depression; echocardiographic findings in the form of LV regional wall motion abnormalities; or troponemia and elevated N-terminal prohormone of brain natriuretic peptide (NT-proBNP) [5,48,49].
The exact incidence of cardiac dysfunction in patients with SAH is difficult to be accurately determined given its imprecise definition and the different diagnostic criteria used in different studies. Indeed, if ECG criteria are used, its incidence seems to be in 100% of patients [8]. If echocardiographic criteria are used, it seems that 10%-15% of patients develop global systolic dysfunction with a left ventricular ejection fraction (LVEF) less than 50%, while almost 30% of patients present wall motion abnormalities [50,51]. Predictors of the development of NSC in patients with SAH include higher Hunt-Hess grade on presentation [52], older age [52,53], female sex [53-55], tobacco abuse [52,55], and aneurysms of the posterior circulation [55].
For patients with acute ischemic stroke, the clinical spectrum of cardiac dysfunction in the context of stroke includes ischemic and non-ischemic myocardial injury with cardiac troponin (cTn)-elevation, acute myocardial infarction, heart failure and NSC, ECG abnormalities, arrhythmias, and sudden cardiac death [19] with variable overlap between the aforementioned disease entities [21]. Recent research highlights that the frequency and severity of these functional and structural alterations peak within three days after the stroke onset [26-28] and are proportionally associated with the severity of ischemic stroke and neurological deficits [34]. Moreover, experimental and clinical studies suggest that specific parameters such as location of ischemic injury influence the effect on the cardiovascular system [56,57]. For example, cardiac injury and dysfunction occur more frequently if the ischemic injury is localized within the central autonomic network and, especially, if the ischemic injury affects the insular cortex [58], which has been linked with the development of NSC [33,34].
Despite considerable inconsistencies in the reporting criteria, acute ECG alterations are more frequently associated with intracerebral hemorrhage or SAH than acute ischemic stroke being additionally more common among patients with troponemia [59,60]. The two major categories of ECG changes during neurogenic heart disease are dysrhythmias (e.g., atrial and ventricular tachyarrhythmias, sinus bradycardia and tachycardia) and repolarization changes [1,2]. The most common repolarization changes are seen in the anterolateral and inferolateral leads [2], including prolongation of the QT-interval and changes in ST-T [61]. In patients with NSC, the most frequent ECG findings are prolongation of the QT-interval and T-wave inversion [62].
Moreover, several studies have reported elevated levels of cTn—specific biomarkers of myocardial injury—and NT-proBNP. These follow acute and severe brain injury, as for example SAH and acute ischemic stroke [63]. With highly sensitive analyses, elevations in cTn can be observed in 30%-60% of ischemic stroke patients [64] and in 20%-40% of patients after intracerebral hemorrhage and SAH with or without coronary symptoms [65]; their levels are correlated with clinical severity of aneurysmal SAH and are associated with persistent ECG and structural echocardiographic alterations, as for example QTc-prolongation, ventricular tachycardia/fibrillation, decreased ejection fraction (EF), and regional wall motion abnormalities [1,66]. Increased troponin levels may be observed in 30% of patients with TBI, and are also associated with a worse prognosis [1]. Furthermore, a variety of studies have reported elevated levels of NT-proBNP in patients with ischemic stroke, SAH, and TBI—conditions that are also correlated with the severity of the disease [50,67-70]. Brain natriuretic peptide (BNP) has potent natriuretic and vasodilator actions, and can decrease cerebral blood flow (CBF), cause hyponatremia, and be elevated in patients with SAH, who develop delayed ischemic injury due to vasospasm [70]. Additionally, neuroscience literature indicates that elevated levels of BNP are associated with troponemia, wall motion abnormalities, diastolic dysfunction, depressed LVEF, and pulmonary edema [54]. However, BNP is a neurohormone, that is not only secreted in the ventricles in response to volume and pressure overload, but also in the brain—specifically in the thalamus, hypothalamus, brain stem, and cerebellum [71-73].
Cardiac systolic impairment is not infrequent after severe neurological injury as it appears in 2% to 30% of patients with SAH [74], in 13% to 29% of patients with ischemic stroke [75], and in 22% of patients with TBI [76]. Furthermore, echocardiographic abnormalities include diastolic dysfunction [76], regional wall motion abnormalities [77], and NSC [7]. The most commonly observed alterations in the echocardiographic patterns are of the apical type; in 25% of patients, there is right ventricular involvement [62] and dysfunction of the basal and midventricular segments [61].
Potential secondary brain alterations in patients with NSC
NSC is a specific form of TTC and a well-documented complication in patients with severe ABI [19,78]. There is growing evidence of brain and neural changes in patients with NSC, which could exacerbate pre-existing cerebral damage. For many years, researchers have recognized that brain activation is implicated in the pathophysiology of TTC [24,79]. Indeed, in a recent study using brain single photon emission computed tomography to assess CBF, Suzuki et al. [24] reported significant increases in CBF in the hippocampus, brainstem, and basal ganglia, with an associated CBF decrease in the prefrontal cortex; these findings may partially explain the brain pathophysiology associated with TTC. As TTC may be protracted [24], it is not surprising that hyperperfusion of the hippocampal and basal ganglia can progress to pathological elevation of neural activity and neuroinflammation. In addition to these findings, Klein et al. [80] have described homogenous structural and functional alterations in brain regions that are mainly associated with cardiac control, emotional processing, as well as cognitive and sensorimotor functions—suggesting communication between the heart and the brain. In a similar vein, studies used magnetic resonance imaging to compare the limbic system in TTC patients and healthy controls report structural and connective differences in patients, which may be associated with dysfunction of the autonomous nervous system, emotional processing, and cognition [81,82]. However, it remains still unclear whether these anatomical alterations are the cause or the consequence of cardiac dysfunction. Templin et al. [81] have recently compared 15 TTC patients and 39 healthy controls by means of functional MRI and found that TTC patients showed hypoconnectivity of the central brain regions of the limbic network. This may indicate that autonomic-limbic integration has a role in the pathophysiology of TTC and even in its specific variant NSC, which could result in dysfunction of emotional processing and of the autonomous nervous system.
Patients with NSC may present symptoms of acute heart failure as a result of severe myocardial dysfunction and injury that can lead to moderate to severe LV dysfunction [1]. On the other hand, there is increasing evidence that cognitive impairment is frequent in patients with heart failure [83]. Indeed, the term “cardiogenic dementia” has been employed to describe the co-existence of “heart and brain failure” after heart disease [84]. Nevertheless, the exact mechanisms of this relationship remain poorly understood and several pathophysiological associations have been proposed, including systemic hypotension with subsequent poor brain perfusion and occult cardiogenic emboli [85,86], as it is present in patients with either reduced or preserved EF [10].
Pathophysiology of secondary brain injury in patients with NSC
Cerebral hypoperfusion
Despite its small size, the brain is a highly metabolic organ, that accounts for about 20% of the body’s consumption of oxygen, while it receives 15% of the total cardiac output [87-90]. Thus the brain is vulnerable to cardiac dysfunction, which could ultimately lead to impairment of cerebral flow. On the other hand, however, heart failure is a systemic and heterogeneous disease associated with fluctuations in blood flow and blood pressure, and may lead to hypoperfusion of peripheral organs and systems, including the brain [78]. It is important that a growing body of evidence supports the role of cerebral hypoperfusion in the development of cognitive impairment in patients with low cardiac output [91-93]. More specifically, experimental and clinical data indicate that brain hypoperfusion results in energetic and metabolic dysregulation, loss of cholinergic receptors, abnormalities in protein synthesis, degeneration of brain capillaries, and neuronal damage that extends from the hippocampus to the temporo-parietal cortex [94]. Moreover, brain hypoperfusion also has a notorious effect on the volume of gray matter—probably due to the latter’s high metabolic demands—and is associated with dysfunction of various cortical areas, resulting in cognitive deterioration [95]. These findings are consistent with the hypothesis of a “head to heart” link in patients with Alzheimer`s disease and support the connection between cerebral hypoperfusion and cognitive impairment [96].
As mentioned previously, the brain receives 15% of cardiac output [89,90], is highly vascularized, and consumes about 20% of the total oxygen budget [96]. Under normal conditions, CBF is approximately 50 mL/min/100 g brain tissue and remains stable within a wide range of mean arterial pressure (MAP), by means of a variety of vascular and neurogenic factors, which ensure reliable autoregulation of the cerebrospinal fluid [10]. However, this homeostatic mechanism is impaired or absent in severe ABI, which can lead to cerebral hypoperfusion, and this may reach ischemic levels and exacerbate primary injury [97]. A key principle of managing critically ill patients with ABI is the maintenance of brain homeostasis, which ensures oxygen delivery to the brain by stabilizing CBF [98]. Cerebral oxygenation critically depends on a close interaction between CBF, arterial content of oxygen, and cerebral oxygen metabolism [11]. Under non-pathological conditions, the brain autoregulates its blood flow to maintain a constant flow regardless of changes in central hemodynamics [99]. CBF is directly related to CPP, which is defined as the difference between mean arterial and intracranial pressures (MAP and ICP, respectively) [99]. In healthy individuals with intact homeostatic mechanisms, the values of CPP remain stable between 50-150 mm Hg by increasing resistance when MAP and CPP increases, and decreasing cerebrovascular resistance when MAP or CPP decreases [97]. This homeostatic mechanism is often impaired in patients with ABI, where ICP can be elevated, and perfusion pressure may then change. Thus, the brain becomes vulnerable to systemic hemodynamic changes [11,100]. In patients with ABI, brain swelling and cerebral edema, cerebrospinal fluid circulation, and neuroinflammation could additionally influence autoregulation [11].
Oxygenation disturbances
In patients with ABI, it is well-documented that complications may be associated with the development of NSC [2] and neurogenic pulmonary edema [14], and may be associated with severe hypoxemia. As highlighted previously, arterial oxygen content is a significant regulator of CBF [11]. Moreover, carbon dioxide (CO2) levels are of crucial importance in maintaining cerebral autoregulation [101], especially in mechanically ventilated patients with ABI [14,102]. It has also been shown that CO2 levels fluctuate in patients with acute heart failure, which may lead to vasoconstriction or vasodilatation of cerebral arteries [103,104]. This point is further supported by findings showing that acute hypoxemic hypoxia (i.e., in seconds to hours) may cause cerebral vasodilation and reduce cerebral resistance, thus leading to an increase in CBF. However, vascular reactivity is additionally affected by hypoxia-induced chemoregulation, as indicated by studies showing that compensatory hyperventilation could lead to hypocapnia, cerebral vasoconstriction, and a drop in CBF [101]. Moreover, a synergistic effect has been observed between hypoxemia and hypercapnia, which is associated with the potent vasodilating properties of CO2 [11].
Taken together, the pathophysiological mechanisms influence CBF through hypoxemia, and changes in blood gas are complex and multifactorial. Even though a detailed examination of all underlying mechanisms related to the role of hypoxemia and hypercapnia on brain function falls outside the scope of the present review, recent high-quality reviews are currently available on the specific topics [11,97,101].
Neurohormonal axis
In recent decades, it has been convincingly demonstrated that activation of the HPA axis is a key component of compensatory anti-inflammatory mechanisms in critically ill patients and involves acute elevation of free cortisol and suppression of the adrenocorticotropic hormones [105,106]. Moreover, there is mounting evidence indicating that this neurohormonal activation plays a pivotal role in the interaction between severe heart disease, structural brain changes, and functional alterations, such as impaired cognition [10]. Indeed, higher levels of cortisol have been found in patients with heart failure than in healthy controls [107]. This is further supported by evidence showing that the incidence of depression and cognitive impairment is significantly higher in heart failure patients than in healthy adults [108]. Moreover, clinical trials in healthy adults demonstrate that even transient hypercortisolism may lead to cognitive impairment, reversible decreases in verbal declarative memory, and altered spatial thinking tasks [109,110]. On the other hand, prolonged exposure to high cortisol levels can cause lasting alterations and atrophy of specific brain regions, through decreased neurogenesis and increased oligodendrogenesis [111]. In addition, stress-induced hypercortisolism may disrupt the plasticity and structural integrity of the hippocampus and the prefrontal cortex, and thus contribute to long-lasting impairments in memory and cognition in critically ill patients [106]. Lastly, high glucocorticoid levels may cause neural damage through activation of systemic inflammatory responses and exacerbation of neuroinflammation [106].
The role of inflammation
Systemic elevation of inflammatory cytokines and oxidative stress have been implicated in the pathophysiology of brain dysfunction after heart disease (Figure 1) [112,113]. Experimental studies in animal models of acute myocardial dysfunction indicate that neuroinflammation may develop after acute heart injury and suggest that immune activation interconnects heart and brain dysfunction [114]. Moreover, it has been convincingly demonstrated that several heart diseases (e.g., myocarditis, myocardial infarction, and heart failure) are associated with activation of inflammatory and immune responses triggered by myocardial injury [95,115]. Moreover, impaired cardiac function may be associated with cerebral hypoperfusion that exacerbates brain inflammatory reactions and excites the cardiac sympathetic system, and this may be related to the development of cognitive impairments in heart failure patients [116]. These findings are further supported by several experimental studies. For example, Hong et al. [117] reported marked cognitive impairment three months after induction of heart failure in a mouse model. This was accompanied by enhanced expression of toll-like receptor-4, tumor necrosis factor (TNF)-α, and IL-6 in the cortex and hippocampus of the animals. Moreover, additional evidence for the development of brain dysfunction in heart failure patients is provided by clinical studies which have found high levels of TNF-α and IL-6 in heart failure patients with cognitive impairment and depressive disorders in comparison to heart failure patients without cognitive dysfunction [118,119].
There is currently increasing interest in the pathophysiological roles of TNF-α in the central nervous system and in the development of neuroinflammation. This role is supported by findings showing that several neurological disorders are associated with marked de novo production of TNF-α [120], which is considered to be one of the main regulators of the brain’s proinflammatory response, and which causes cell dysfunction and death through glutamate-mediated neurotoxicity [120]. Moreover, it is widely accepted that IL-1β is a hallmark of neuroinflammation, which strongly contributes to cognitive impairment [121]. Furthermore, TNF-α and IL-1β can potentiate marked production of IL-6, which is also associated with neurodegenerative processes and cognitive dysfunction [122]. In addition, cardiac oxidative stress, activation of cascades, and release of proinflammatory cytokines (e.g., TNF-α, IL-1β, and IL-6) have been consistently described in patients with ischemic heart disease and in heart failure patients [10,123]. It is now known that IL-6 receptors are located in crucial areas, such as the hippocampus and the cerebral cortex. Therefore, it is not surprising that IL-6 may be an important factor in neuroinflammation, neuronal damage, and apoptosis, with subsequent development of severe neuropathological processes, so this offers a promising therapeutic target [10,124].
The situation is further complicated if it is born in mind that activation of inflammatory cascades and oxidative stress may induce neuropathological processes due to disruption of the BBB and activation of glial cells [12,13]. Systemic inflammation may cause alterations in the structure and function of BBB, which may be disruptive or non-disruptive [13,87]. Disruptive BBB alterations include changes at the histological level, such as destruction of endothelial cells, and include molecular changes leading to endothelial apoptosis, membrane abnormalities, mitochondrial damage, and modification of tight junctions [13]. Non-disruptive alterations may be due to molecular changes [13]. Many cytokines and inflammatory factors, for example, TNF-α and IL-1β, have been found to mediate some of these changes (e.g., cerebral endothelial activation associated with neuroinflammation), and increased permeability of the BBB [125]. Moreover, inflammation associated with ischemic myocardial injury may promote activation of astrocytes [126]. Besides its effects on astrocytes, inflammation also plays a role in leukocyte recruitment across the BBB. Thus, migration of monocytes to the central nervous system promotes their differentiation into microglia, which has also been associated with neuroinflammation [127]. It should be remembered that activated microglia upregulate the production of proinflammatory cytokines (e.g., IL-1, IL-6, and TNF-α), thus contributing to the development and progression of neurodegeneration [128].
Finally, critical illness such as ABI may lead to excessive elevations of circulating glucocorticoids, which may generate or exacerbate inflammation-induced hippocampal and prefrontal cortical damage [106].
Ischemic stroke in patients with NSC
Myocardial dysfunctions, such as NSC, are well-established complications of severe brain injury [4,129]. On the other hand, TTC is associated with acute and severe LV dysfunction, which could contribute to LV thrombus formation—with potential cardioembolic complications, such as cardioembolic stroke [15]. The incidence, however, of LV thrombus formation and related embolic complications varies between studies [15,130,131]. In a systematic review of 26 clinical studies, de Gregorio [15] reported that LV thrombus formation occurred in about 5% of patients with TTC, and that 25% of the whole study population develop major stroke or transient ischemic attack. Additionally, a recent multicenter study, which enrolled 1,676 patients with TTC, reported that intraventricular thrombus formation and/or embolism was present with a prevalence of 3.3% and that 23.2% of these patients developed ischemic stroke [131].
Thromboembolic events in TTC are probably related to the depressed LV systolic dysfunction and the subsequent blood stasis during the acute phase [88,131-133]. Moreover, endothelial dysfunction, hypercoagulability [131,134], activation of inflammatory pathways, and release of cytokines may provide additional pathophysiological mechanisms [131,135]. Indeed, hypercoagulable state associated with NSC underlies the Virchow triad (e.g., endothelial cell damage, blood stasis, and platelet activation) [136]. Possible pathomechanisms associated with hypercoagulability in TTC include the catecholamine storm during the acute phase of the disease with subsequent increased platelet activation and aggregation [137]. Furthermore, previous research indicates that inflammatory milieu with elevated plasmatic levels of IL-6 and IL-7 is an integral part of LV dysfunction [138,139]. Moreover, several studies have revealed higher rates of LV thrombus formation and, thus, associated cardioembolic events, in patients with lower EF, right ventricular involvement, apical ballooning, and temporal anterior wall dysfunction in takotsubo syndrome [20,140,141]. The hypothesis of LV thrombus formation with higher stroke rates related to apical ballooning is further supported by a recent retrospective study, which examined clinical and imaging data of 72 patients hospitalized with takotsubo syndrome, and detected apical ballooning with anterior wall motion abnormalities in 90% of the patients. Moreover, these patients showed significant recovery of LV function within two months of the event. The authors proposed that rapid changes in LV morphology might be a complementary mechanism contributing to LV thrombus formation and suggested that oral anticoagulation should be evaluated if not contraindicated until recovery of ventricular function [142].
Additionally, a variety of dysrhythmias have been observed in patients with TTC, including supraventricular tachyarrhythmias, ventricular tachycardia, ventricular fibrillation, asystole, and pulseless electrical activity [143]. More specifically, patients with TTC are at increased risk of developing various atrial arrhythmias like atrial fibrillation due to temporal left atrial dysfunction during the acute phase [144]. The prevalence of atrial fibrillation in patients with TTC is high, reaching a percentage of 25% [144,145] compared to 1% in the general population [146]. Moreover, its presence in TTC patients serves as a powerful predictor of short-term and long-term mortality [144,145]. However, it is conceivable that the number of unreported episodes of paroxysmal atrial fibrillation among patients with TTC is higher due to the fact that many of them have a “silent” clinical presentation [147] making its diagnosis a clinical challenge.
Finally, despite that stroke in patients with TTC is primarily thought to be associated with LV thromboembolism [88,132,133], the question arises whether pre-existing cardiovascular risk factors are relevant to the development of stroke in patients with TTC. Moreover, several studies have shown an increased rate of common cardiovascular comorbidities and atherosclerotic risk factors in patients with TTC, including hypertension, diabetes mellitus, hyperlipidemia, and smoking [148,149]. Indeed, a recent retrospective study of 27,970 patients hospitalized with TTC examined the trends in the incidence of ischemic stroke stratified by subtype (cardioembolic vs. thrombotic). The authors reported that thrombotic etiology of ischemic stroke was more common among patients with TTC and hypothesized that the higher rates of risk factors among these patients contribute significantly to the higher incidence of stroke of thrombotic etiology [149].
Despite the fact that published data are scarce, cardioembolic complications, such as ischemic stroke, could additionally develop in patients with cardiac dysfunction—after primary acute neurological injury—making the management of these patients a therapeutic challenge.
Clinical management and therapeutic implications
Screening for high-risk patients
Early detection of cardiovascular complications caused by ABI has become paramount. In fact, the therapeutic goal in patients with ABI is to ensure brain homeostasis and oxygenation, avoid intracranial hypertension, and decrease oxygen consumption [49,150,151] in order to minimize secondary brain insults [152]. Moreover, as previously discussed, ABI may be complicated by the development of NSC, which impairs LV function and systemic arterial pressure, leading to cerebral hypoperfusion, impaired brain homeostasis, and reduced oxygen supply to the brain [49,153,154].
Hence, close hemodynamic monitoring not only leads to improved systemic and cerebral hemodynamics, but also plays an additional fundamental role in avoiding secondary ischemic events. Mutoh et al. [153] compared the usefulness of transpulmonary thermodilution, a less invasive technique for hemodynamic monitoring, with pulmonary artery catheter monitoring in 16 SAH patients diagnosed with vasospasm. The authors then reported reduced frequencies of vasospasm and cardiopulmonary complications in patients for whom goal-directed therapy was guided by transpulmonary thermodilution. In addition, a multicenter prospective cohort study of hemodynamic variables using thermodilution in patients with poor grade SAH demonstrated elevated indexes of lung water and pulmonary vascular permeability, together with a systemic vascular index corresponding to afterload mismatch and resembling heart failure [154].
For patients with stroke, myocardial injury is often detected and strongly associated with poor outcomes. The term “stroke-heart syndrome” involves a broad spectrum of cardiac complications after ischemic stroke, such as arrhythmias, cardiac injury and dysfunction [64], and NSC [155]. Early blood pressure and continuous ECG monitoring is suggested in order to be able to identify ischemic stroke patients at high risk of cardiac complications at an early stage. Moreover, as extensive data have shown a relationship between LVEF and stroke severity and an association with neurological disability [156], we strongly recommend early echocardiographic monitoring in severely affected patients (National Institutes of Health Stroke Scale [NIHSS] score >10), patients with clinical signs of myocardial dysfunction and with elevated cardiac biomarkers, and patients with disturbances during monitoring such as arrhythmias and severe hypertension [44]. Furthermore, clinicians should also take into account the likely effect of atrial fibrillation, of which incidence has been found to be statistically significantly higher in both, the group of patients with severe stroke and in patients with impaired systolic function [156]. Finally, prolonged cardiovascular monitoring is recommended in high-risk patients with right-located stroke, especially those with insular involvement, older patients, and patients with a history of hypertensive or coronary artery disease, or with intense emotional stress [2].
Finally, in patients with NSC, a single bed-side risk score including several parameters, as for example lower LVEF, apical type, elevated troponin levels, increased inflammatory biomarkers, and prior history of vascular disease, may help identify patients at a higher risk of adverse events (e.g., intraventricular thrombus formation and embolism) and, thus improve outcome [131].
Therapeutic strategies
NSC is frequently managed conservatively due to its reversible nature. In patients with classic uncomplicated TTC, the most appropriate and beneficial therapeutic intervention is the combination of β-blockers with angiotensin-converting enzyme (ACE) inhibitors [157]. Indeed, several studies demonstrate the therapeutic benefits of β-blockers, as they lead to rapid recovery of LV function and inhibition of the Gs-protein metabolic pathway, while ACE inhibitors prevent vasospasm and reduce hypertension [158-160]. Additionally, previous research data demonstrate that pre-existing treatment with β-blockers is associated with milder stroke severity due to their sympatholytic action, anti-inflammatory properties, and reduction of norepinephrine-stimulated myocytes apoptosis [161,162]. Moreover, a classical study of Neil-Dwyer et al. [163] assessed the cardioprotective effect of propranolol plus phentolamine in patients with SAH and reported reduced myocardial necrotic lesions in patients receiving the combination propranolol/phentolamine when compared to the placebo group. These findings are in accordance with studies examining the effects of β-blocker in TBI patients with a recent meta-analysis of nine studies showing a significant reduction in in-hospital mortality in adult patients with TBI [164]. Similarly, a multicenter, prospective, observational study in patients with TBI found that the administration of β-blockers after TBI was associated with lower mortality and that propranolol showed better effects than other β-blockers [165]. In contrast, in a recent meta-analysis of three retrospective studies, which included 691 patients with SAH, no benefit of pre-admission β-blocker on mortality or on cardiac dysfunction, or vasospasm was observed [166]. NSC with persistent LV dysfunction should be treated according to heart failure guidelines, including ACE inhibitors, angiotensin receptor blockers, and β-blockers [167]. However, β-blockers are contraindicated in patients with decompensated heart failure [142]. Further research is needed before β-blockers and ACE inhibitors can be routinely recommended to patients with NSC due to acute ischemic or hemorrhagic stroke.
Maintenance of systemic hemodynamics is crucial for patients with ABI, in order to ensure brain homeostasis, adequate CPP, and accurate brain oxygenation and, thus, to minimize or even avoid secondary brain injury [49,154,168]. Despite the general agreement that vasopressors should be avoided in patients with TTC, a retrospective study reported a similar total mortality between SAH patients with NSC and SAH patients without NSC [169]. However, due to the pathophysiologic background of the disease, it is generally recommended that adrenaline, noradrenaline, dobutamine, milrinone, and isoproterenol should be avoided in patients with TTC secondary to neurological illness [170]. On the other hand, if indicated, the use of the inotropes dobutamine and milrinone may improve cardiac function in SAH patients with NSC, with patients with cerebral vasospasm being especially benefited by milrinone [8,170]. Patients with cardiogenic shock without LV outflow tract obstruction (LVOTO) usually present with hypotension. For the specific patient group, the use of dobutamine/dopamine is recommended for its treatment due to their positive inotropic effect. Inotropic support should be discontinued if the patient develops moderate or severe LVOTO. In case of refractory hypotension with impaired internal organ blood supply, vasopressor administration should be considered [157,171]. If cardiogenic shock is accompanied by LVOTO, the treatment of choice is combined with fluid administration of β-blockers; phenylephrine administration may also be considered in preference to positive inotropic drugs (e.g., norepinephrine) [157,171-173]. Because of its cardioprotective action and increase in blood supply to internal organs and intra-aortic balloon pump, the use of levosimendan should be evaluated in patients with refractory SAH-associated cardiogenic shock [8,174,175].
More clinical trials are required in order to indicate the optimal therapeutic management in patients with concurrent ABI and NSC and, consequently, improve their cardiac function and minimize secondary brain injury. More specifically, further research examining the exact role of β-blockers and ACE inhibitors is strongly recommended. Moreover, clinical trials should evaluate other therapeutic alternatives, as for example estradiol administration in postmenopausal women with NSC or medications, which are found to be effective in the management of patients with heart failure (e.g., angiotensin-neprilysin receptor inhibitors and mineralocorticoid receptor antagonists). Finally, more experimental and controlled clinical studies are needed to determine the effectiveness of alternative therapeutic options, such as targeting inflammation and coronary microcirculation.
Conclusions
In recent years, it has been convincingly demonstrated that ABI, such as SAH, intracerebral bleeding, ischemic stroke, and TBI may cause severe cardiac complications. The pathophysiology of these brain-heart interactions is complex and involves sympathetic hyperactivity, activation of the HPA-axis as well as immune and inflammatory pathways. Patients with neurogenic cardiac dysfunction are likely to develop delayed secondary cerebral complications associated with these heart complications. Still, however, little is known about the pathophysiology of this phenomenon. The aim of the current review was to highlight the main pathophysiological mechanisms of stroke-heart-brain interactions and cardiac-associated secondary brain complications in patients with concurrent ABI and especially in patients with stroke and NSC. As was demonstrated, NSC induces severe systemic and cerebral inflammation, activation of the HPA axis, BBB dysfunction, and hemodynamic compromise, so neurocritical care patients are vulnerable to the development of delayed secondary cerebral events. The therapeutic management of neurogenic cardiac dysfunction is conservative, primarily oriented to the improvement of cardiac function, prevention of further neuroinflammation, provision of adequate oxygenation, and maintenance of brain homeostasis and CPP. Further studies are needed to investigate the complex pathophysiological interactions between heart and brain.