Introduction
Moyamoya disease (MMD) is characterized by a progressive stenosis at the terminal portion of the internal carotid artery (ICA) and an abnormal vascular network at the base of the brain.1,2 The etiology of MMD remains unknown, but recent advances have been made in understanding the molecular biology and pathophysiology of this rare entity. Previous studies explored genetic factors and revealed several loci associated with moyamoya disease; 3p24-p26,3 6q25,4 8q23,5 and 17q25.6 More recently, the RNF213 gene (RNF213) in the 17q25-ter region was identified as a novel susceptibility gene for MMD among East Asian population.7,8,9 A polymorphism in c.14576G>A in RNF213 was identified in 95% of familial patients with MMD and 79% of sporadic cases,7 and RNF213 was found to correlate with the early-onset and severe forms of MMD, indicating its value as a good biomarker for predicting prognosis.10 The exact mechanism by which the RNF213 abnormality relates to moyamoya disease remains unknown, but recent reports using genetically engineered mice lacking RNF213 by homologous recombination provide new prospects for the basic research of this rare entity.11,12 In this review article, we focus on the genetics and biomarkers of MMD, and sought to discuss their clinical implication.
Basic pathology of MMD
Histo-pathological characteristics
Intimal hyperplasia and medial thinness are well-known histo-pathological characteristics of MMD.2,13,14 The thickened intima has increased number of smooth muscle cells, which are thought to be synthetic-type smooth muscle cell migrating from medial layer.15 Degradation of the smooth muscle cells in the medial layer and subsequent death of the vascular smooth muscle cells are thought to result in the characteristic finding of medial thinness not only at the terminal ICA but also at the peripheral middle cerebral artery.14 Together with the waviness and duplication of the internal elastic lamina, intracranial arteries of moyamoya disease generally have intrinsic fragility, which should also be mentioned as one of the pitfalls of revascularization surgery while manipulating peripheral middle cerebral artery during extracranial-intracranial (EC-IC) bypass.16
Macroscopic pathology of MMD; physiological reorganization system
For the better understanding of MMD as dynamic disease, it is particularly important to revisit Suzuki's angiographic staging,1,2 which represent the natural course of the angio-architecture in moyamoya patients by physiological reorganization system. Suzuki's angiographic staging does not reflect the severity of MMD, while it explains how 'moyamoya disease' compensates its ischemic condition by physiological process.16 At the early stage of MMD, progressive stenosis of the terminal ICA occurs while it is subsequently compensated by abnormal vascular networks at the base of the brain at stage III. In substantial number of patients, ischemic brain is further compensated by trans-dural anastomosis from external carotid system without surgical intervention at stage IV to VI, resulting in the disappearance of moyamoya vessels. We named this physiological reorganization process as 'internal carotid (IC)-external carotid (EC) conversion'.16 Concept of revascularization surgery for MMD, either direct or indirect revascularization, is to accomplish successful 'IC-EC conversion' to prevent insufficient reorganization resulting in cerebral infarction. Therefore, the concept of revascularization surgery for MMD should be based on the idea to support the intrinsic compensatory nature of MMD, rather than to eradicate the pathophysiology of this entity.
Genetics of MMD
Background and previous reports
Since some patients with MMD show autosomal dominant inheritance pattern, genetic factors have been implicated in the etiology of MMD. In fact, gene loci have been identified in 3q24-p263 and 8q235 in genome-wide analysis, and in 6q254 and 17q256 in a chromosomal search for familial MMD. The RNF213 in the 17q25-ter region was more recently identified as a susceptibility gene for MMD among East Asian population.7,8,9 We found that a polymorphism in c.14576G>A in RNF213 was identified in 95% of familial patients with MMD and 79% of sporadic cases.7 Miyatake et al. reported that patients with the polymorphism of c.14576G>A in the RNF213 gene had significantly earlier onset and a more severe form of MMD, such as the presentation of cerebral infarction and posterior cerebral artery stenosis.10 To investigate the potential role of RNF213 polymorphism, Liu et al.8 investigated the effects of RNF213 suppression on zebra-fish vasculature by generating RNF213 knockdown zebra-fish, demonstrating that zebra-fish lacking RNF213 showed severely abnormal sprouting vessels in the head region, especially from the optic vessels. It was suggested that RNF213 is involved in a novel signaling pathway in intracranial angiogenesis.8
Generation of RNF213-deficient mice (RNF213-/-)
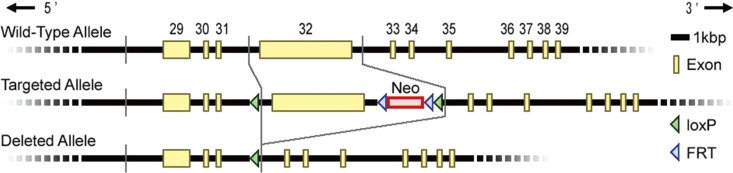
Using Cre-lox system, we generated RNF213-deficient mice with C57BL/6 background by deleting exon 32 of RNF213, which is the largest exon in the RNF213 gene (Figure 1).12 Finally, heterozygous male and female mice were bred to produce homozygous offspring (homozygous knockout mice, RNF213-/-). Genotyping was performed by PCR using specific primers to exon 32 (forward; 5'-CTCAGTGGTGGTGTTGGATG-3', reverse; 5'-CTCTTTCTCGTTGGGACTGC-3') and the deleted allele (forward; 5'-ATAACCTCAGGCACCAATCG-3', reverse; 5'-TCCCTCTAGGCAGGAAGGAT-3').12 The complete removal of RNF213 exon 32 from genomic DNA was confirmed in RNF213-/- (Figure 2A). Both homozygous mutant (RNF213-/-) and heterozygous mutant mice (RNF213-/+) were born and grew normally.
Temporal changes of vascular anatomy in RNF213-/-
Then we performed magnetic resonance angiography (MRA) using a dedicated small animal scanner with a 50-mm bore operating at 9.4 Tesla field strengths (AV400WV, Bruker BioSpin).12 A high resolution three-dimensional gradient-echo time-of-flight MRA sequence was used to acquire MRA images. As a result, no significant difference was observed in the MRA findings of cervical/intracranial arteries between RNF213-/- and wild-type littermates (Wt.) from 32 to 64 weeks of age.12 The anatomy of the circle of Willis was evaluated by a trans-cardiac injection of carbon black dye in RNF213-/- and Wt. (Figure 2B, C). No significant difference was observed in the structure of the major arteries at the base of the brain between RNF213-/- and Wt. at 16 weeks of age (Figure 2B, C). Vessel diameter was sustained at 40 weeks of age in RNF213-/-, and no steno-occlusive changes were observed around the terminal portion of the internal carotid artery. An abnormal vascular network did not develop at the base of the brain in RNF213-/-.12
Modification of vascular remodeling in RNF213-/- after carotid artery ligation
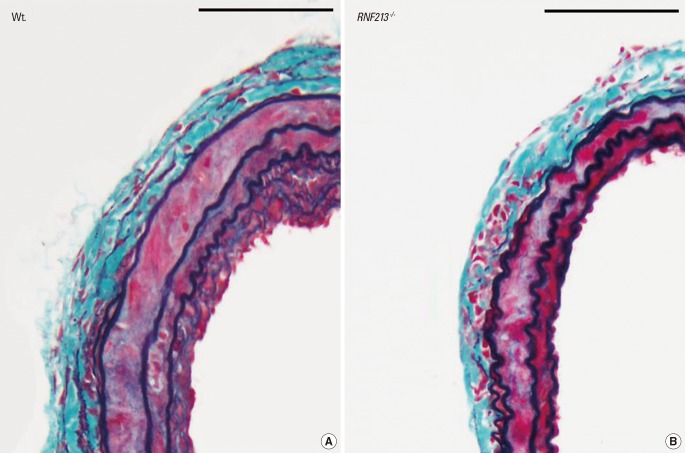
We also evaluated the histo-pathological characteristics of the vascular wall structure in RNF213-/-, and showed no apparent abnormality in mutant mice including intimal hyperplasia or medial layer thinness, both of which are the characteristic findings of MMD. Thus we employed common carotid artery (CCA) ligation model, which reproducibly induces arterial wall hyperplasia.17 After CCA ligation, Wt. showed temporary hyperplasia of the intima and medial layers (Figure 3A), which was consistent with previous reports;18 however, RNF213-/- did not exhibit temporary hyperplasia of the intima and medial layers at the same time point (Figure 3B). Both the intima and medial layers were significantly thinner in RNF213-/- than in Wt. 14 days after CCA ligation, while no significant difference was observed in vascular wall thickness 7, 21, and 28 days after CCA ligation.12
Role of RNF213 polymorphism in MMD
Our results demonstrated that RNF213-/- grew normally, and no significant difference was observed in MRA findings or the anatomy of the circle of Willis between RNF213-/- and Wt. under normal conditions. The histo-pathological characteristics of the vascular wall structure in RNF213-/- showed no apparent abnormality, thus it is conceivable that an abnormality in RNF213 does not sufficiently induce MMD. Recently, Kobayashi and colleagues alternatively generated RNF213-/- and suggested that target disruption of RNF213 improved glucose tolerance by protecting inlet β cells when they are mated with Akita diabetic mice, but they also failed to detect any cerebrovascular pathology mimicking MMD,11 which is consistent with our result.
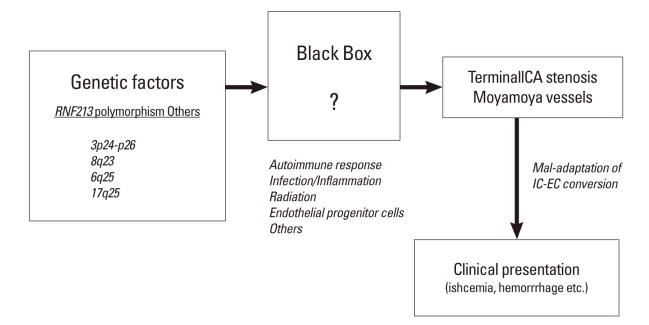
Although it is still undetermined whether the polymorphism of c.14576G>A in patients with MMD is a loss of function mutation or a gain of function mutation, the initial report of knockout zebra-fish by Liu et al.8 suggested that it could be a loss of function mutation. In our study, we did not observe any evidence of the characteristic findings of MMD in RNF213-/- under normal physiological conditions until 64 weeks of age. However, following CCA ligation, which reproducibly induced temporary hyperplasia of the adjacent vascular wall,17,18 we showed that the medial layer of CCA was significantly thinner in RNF213-/- than in Wt. 14 days after CCA ligation, which matched one of the histological characteristic findings of MMD.2 Therefore, RNF213 deficiency could lead to vascular fragility including medial thinness, which may make vessels more vulnerable to hemodynamic stress and secondary insults, which facilitates the development of MMD. Furthermore, it is conceivable that secondary insults in addition to RNF213 abnormality, such as an autoimmune response,19,20 infection/inflammation, and radiation2 may be necessary for the development of MMD. In fact, the incidence rate of MMD was shown to be as low as 0.53 per 100,000 population in Japan,21 while 1% of the Japanese population carries the polymorphism of c.14576G>A in the RNF213 gene,8 again suggesting the importance of additional insult to induce MMD. This observation could be illustrated as 'two-hit theory', as is also true in a variety of disorders (Figure 4).
Possibility of medial thinness as an initial histo-patholohical change in MMD
Autopsy studies of MMD have shown that the outer diameter, as well as intra-luminal diameter, of the affected arteries of the circle of Willis is smaller in MMD.22 Intraoperative finding also suggested smaller outer diameter of the terminal ICA in MMD.23 More recently, the arterial constrictive remodeling with early narrowing of outer diameter of the ICA was proposed to be the intrinsic pathogenesis of MMD. Based on their finding of three-dimensional constructive interference in steady-state magnetic resonance imaging demonstrating that marked narrowing of the outer vascular diameter may precede internal vascular stenotic progression,24 it could be alternatively possible that the thinness of the entire vascular wall in RNF213-/- might represent the very early finding of the constrictive remodeling as a characteristic of MMD.
Possibility RNF213 polymorphism as a gain of function mutation
Alternatively, we should consider another possibility that the RNF213 polymorphism in patients with MMD is a gain of function mutation.25,26 Since RNF213-/- in our study, which represented a loss of function mutation, did not spontaneously develop MMD under normal conditions, it is also conceivable that the polymorphism of c.14576G>A in RNF213 contributes to the gain of function of the RNF213 product in patients with MMD.12 In fact, we obtained conflicting results; the medial layer was significantly thinner in RNF213-/-, which is again the characteristic finding of MMD, but temporary intimal hyperplasia was significantly suppressed in RNF213-/- during vascular remodeling after CCA ligation, which is not a characteristic of MMD. Further studies using knock-in mice carrying the polymorphism of c.14576G>A in the RNF213 gene would address this important issue.
Biomarkers of MMD: clinical implications
Expression of angiogenic factors and pro-inflammatory molecules in MMD
It has been well known that patients with MMD have increased expression of various pro-inflammatory molecules as well as angiogenic factors in serum and/or cerebrospinal fluid (CSF). Increased expression of basic fibroblast growth factor (bFGF) in CSF from moyamoya patients was considered to be specific and was not thought to be simply related to cerebral ischemia.27 The bFGF level was apparently elevated in the patients in whom neovascularization formed from indirect revascularization, suggesting that the bFGF level is a useful indicator to predict the efficacy of indirect revascularization after surgery.27 Nanba et al.28 alternatively reported the increased expression of hepatocyte growth factor in CSF and intracranial artery in MMD. Regarding serum level of these molecules, Kang and colleagues comprehensively investigated the expression of matrix metalloproteinases, interleukins, and growth factors, and they found that patients with MMD exhibited significantly higher plasma concentrations of matrix metalloproteinase (MMP)-9, monocyte chemoattractant protein-1, interleukin-1β, vascular endothelial growth factor (VEGF), and platelet-derived growth factor-BB.29 In light of the recent report by Park and colleagues demonstrating that moyamoya patients with VEGF polymorphism of the CC genotype of VEGF-634 had better collateral vessel formation after revascularization surgery, increased serum level of VEGF in patients with MMD should participate in the induction of pial synangiosis after indirect revascularization surgery as well as to the development of abnormal vascular networks at the base of the brain.30 Alternatively, increased expression of serum MMP-9 may contribute to the spontaneous intracranial hemorrhage in MMD, as well as to the higher risk for cerebral hyperperfusion syndrome after direct revascularization surgery in moyamoya patients.31,32
Pharmacological approach to the biomarkers of MMD: clinical application
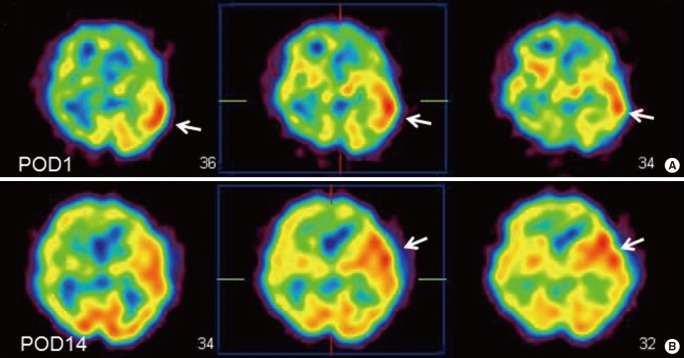
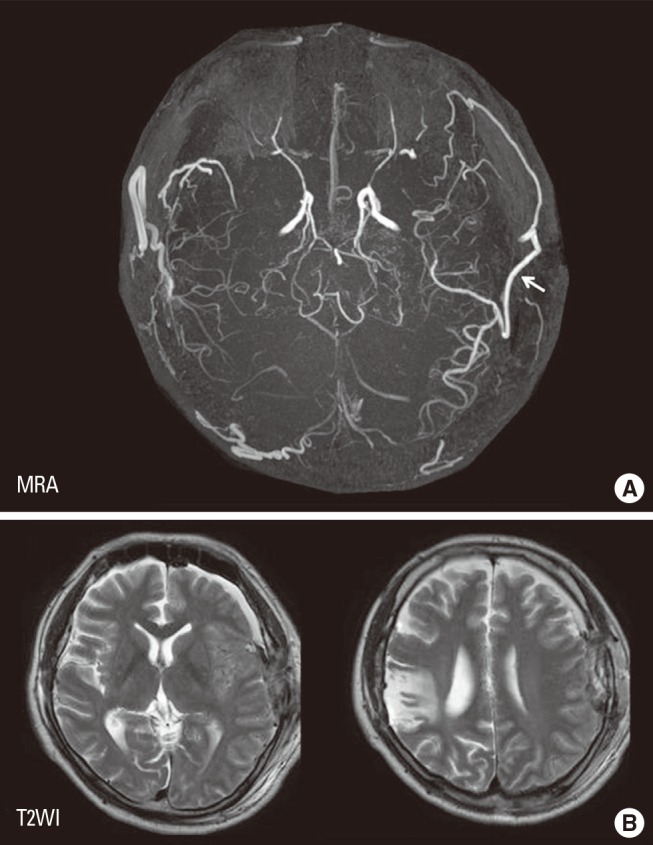
EC-IC bypass is an established management of MMD,2 but cerebral hyperperfusion syndrome (CHS) is one of the critical complications of EC-IC bypass for MMD, which could result in focal neurological deterioration and/or delayed intracranial hemorrhage.32,33,34,35,36 Although the mechanism underlying the higher incidence of CHS among patients with MMD has been undetermined, intrinsic vulnerability of vascular structure including blood-brain barrier was considered to lead to vasogenic edema and hemorrhagic conversion following revascularization to the chronic ischemic brain.31 Since MMP-9 is known to participate in edema formation and hemorrhagic conversion after cerebral ischemia-reperfusion injury,37,38 it was conceivable that the increased expression of MMP-9 may contribute in part to CHS in moyamoya patients. Minocycline hydrochloride was known to play a role in blocking MMP-9,39 and is also known to have a neuroprotective effect against ischemic brain injury,40 thus we attempted to use minocycline perioperatively to prevent CHS.41 The result showed that minocycline significantly reduced the incidence of focal neurological deterioration due to CHS after EC-IC bypass for MMD,41 indicating that increased MMP-9 expression in MMD could participate in the pathology of postoperative CHS. This representative 43-year old man with ischemic-onset MMD underwent left EC-IC bypass, and demonstrated significant focal increase in cerebral blood flow at the site of the anastomosis which prolonged over 2 weeks (Figure 5). He was managed by intravenous administration of minocycline hydrochloride (200 mg/day) under strict blood pressure control, and did not suffer any neurological deterioration during the perioperative period. Postoperative magnetic resonance imaging showed no evidence of vasogenic edema (Figure 6), and he was discharged without neurological deficit. We consider that minocycline has potential role for preventing cerebral hyperperfusion to be symptomatic, by blocking MMP-9 in the acute stage after EC-IC bypass. Future investigation of various biomarkers related to MMD may further improve the outcome of MMD.
Conclusions
Although the etiology of MMD remains unknown, recent advances have been made in understanding the molecular biology and pathophysiology of this rare entity. Genome-wide and locus-specific association studies identified RNF213 as an important susceptibility gene of MMD. Ongoing research using genetically engineered mice lacking RNF213 by homologous recombination may provide new prospects for the basic research of this rare entity. Alternatively, variety of biomarkers are known to be involved in MMD, and an increased expression of angiogenic factors and pro-inflammatory molecules such as vascular endothelial growth factors and MMP-9 could be the therapeutic target in the future.