Lacunar Infarction and Small Vessel Disease: Pathology and Pathophysiology
Article information
Abstract
Two major vascular pathologies underlie brain damage in patients with disease of small size penetrating brain arteries and arterioles; 1) thickening of the arterial media and 2) obstruction of the origins of penetrating arteries by parent artery intimal plaques. The media of these small vessels may be thickened by fibrinoid deposition and hypertrophy of smooth muscle and other connective tissue elements that accompanies degenerative changes in patients with hypertension and or diabetes or can contain foreign deposits as in amyloid angiopathy and genetically mediated conditions such as cerebral autosomal dominant arteriopathy with subcortical infarcts and leukoencephalopathy. These pathological changes lead to 2 different pathophysiologies: 1) brain ischemia in regions supplied by the affected arteries. The resultant lesions are deep small infarcts, most often involving the basal ganglia, pons, thalami and cerebral white matter. And 2) leakage of fluid causing edema and later gliosis in white matter tracts. The changes in the media and adventitia effect metalloproteinases and other substances within the matrix of the vessels and lead to abnormal blood/brain barriers in these small vessels. and chronic gliosis and atrophy of cerebral white matter.
Pathology
Vascular pathology
Two major vascular pathologies underlie brain damage in patients who have important abnormalities involving small size penetrating brain arteries and arterioles; 1) thickening of the arterial media with encroachment on the arterial lumens and 2) obstruction of the origins of penetrating arteries by parent large intracranial artery intimal plaques. Hypertension, diabetes and other as yet undetermined genetic and other factors may promote thickening of the media of penetrating arteries by fibrinoid deposition and hypertrophy of smooth muscle and other connective tissue elements. The arterial media can also contain foreign deposits as in amyloid angiopathy and genetically mediated conditions such as cerebral autosomal dominant arteriopathy with subcortical infarcts and leukoencephalopathy (CADASIL).
Miller Fisher, nearly a half century ago, analyzed the arterial pathology that caused small deep infarcts by carefully assessing serial sections of vessels at necropsy.1 He observed that the penetrating arteries that supplied the territory of lacunar infarcts showed a characteristic vascular pathology. These tiny vessels often contained focal enlargements and small hemorrhagic extravasations through the walls of these arteries. Subintimal foam cells sometimes obliterated the lumens, and pink-staining fibrinoid material was present within the vessel walls. The arteries in some regions were often replaced by whorls, tangles, and wisps of connective tissue that obliterated the usual vascular layers. Fisher called these processes segmental arterial disorganization, fibrinoid degeneration, and lipohyalinosis.1 Figure 1, given to me by Fisher, shows a severely narrowed penetrating artery in a patient with severe hypertension. The media is hypertrophied and contains fibrinoid material.
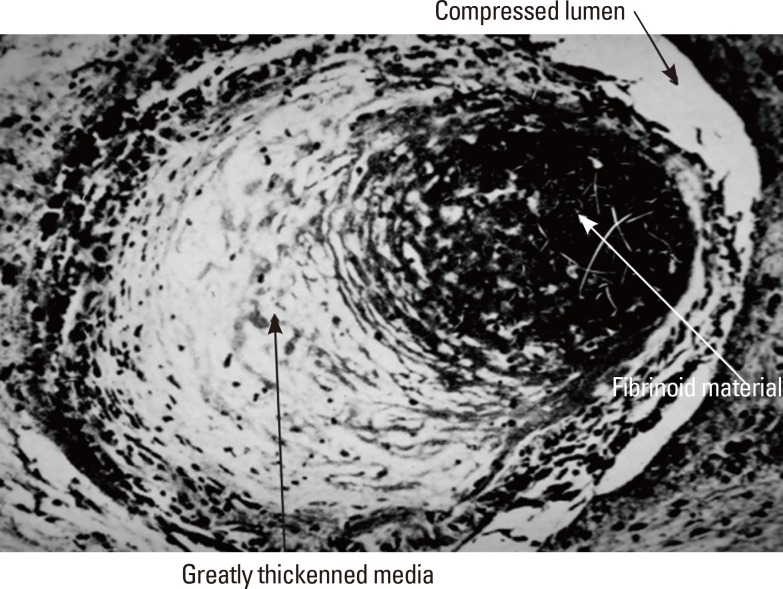
Small penetrating artery showing lipohyalinosis and fibrinoid necrosis (white arrow); lumen is considerably compromised (upper black arrow). The thickened media contains lipohyalinotic material (Lower black arrow) (Supplied by Dr. C. Miller Fisher.).
When Fisher reviewed the charts of 114 patients who had lacunes at necropsy, all but three patients had hypertension defined by a prior history of high blood pressures, elevated blood pressure recorded on examination, or heart weight exceeding 400 g without another explanation.2 The onset of the clinical symptoms of brain infarction often developed at the onset or hypertension or during periods of an increase in blood pressure values. Fisher attributed segmental arterial disorganization and lipohyalinosis to hypertension. At the time of his studies, performed nearly a half century ago, there was little effective treatment for hypertension and hypertensive crises and hypertensive encephalopathy were common. Later epidemiological studies of patients with small deep infarcts has shown that the presence of hypertension, although very common, seemed no different than the frequency in patients with large artery extracranial and intracranial occlusive disease, raising doubt about the crucial importance of high blood pressure in causing the vascular changes that caused these small deep infarcts. The role of diabetes, hyperlipidemia, and aging in causing penetrating artery lesions remains uncertain. Curiously, necropsy examination of patients with chronic small disease does not show evidence of thrombosis or past thrombotic occlusion of these diseased arteries.3
The other major pathology that directly involves the walls of penetrating arteries is infiltration of the coats of these vessels with materials foreign to vessels. The penetrating arteries, arterioles and capillaries in a hereditary condition dubbed CADASIL contain a granular material in the media that extends into the adventitia. This disorder is caused by dominant mutations in the NOTCH3 gene that encodes a transmembrane receptor predominantly expressed in vascular smooth muscle and pericyte cells in adults.4,5 This receptor is needed for the structural and functional integrity of small vessels-small arteries, arterioles and capillaries. Periodic acid-Schiff (PAS) staining suggests the presence of glycoproteins, but staining for elastin and amyloid have been negative. Figure 2 is a PAS stained section that shows glycoprotein material within vessel walls in a CADASIL. The nature of the deposited material within the vessel wall and matrix remains unknown. Smooth muscle cells in the media are swollen and often degenerated. The endothelium may be absent and replaced by collagen fibers. At times abnormalities also found in hypertensive patients are also found, including duplication and splitting of the internal elastic lamina, adventitial fibrosis and hyaline change, and fibrosis and hypertrophy of the arterial media.
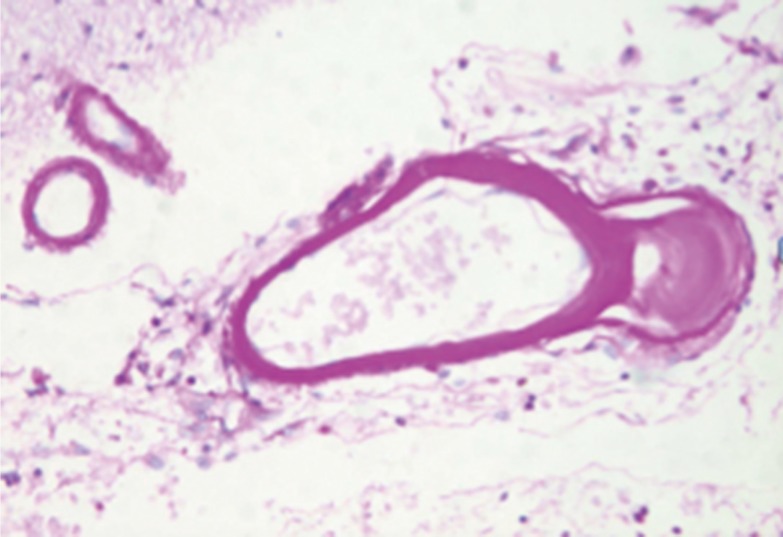
Periodic acid Schiff (PAS) stain showing bright pink staining material within small cerebral arteries in a patient with CADASIL.
Another hereditary angiopathic condition, now known to be associated with mutations in a gene that encodes procollagen type IV alpha 1 (Col4A1), has been identified. This type of collagen is very important in vascular basement membranes. This genetic mutation affects small brain arteries as well as larger retinal and cerebral arteries.5,6
Cerebral amyloid angiopathy (CAA) affects small arteries and arterioles in the leptomeninges and cerebral cortex; involved arteries are thickened by a acellular hyaline material that stains positively with periodic acid-Schiff stains, and has an apple-green birefringence with polarized Congo red stain.7,8 Arteries and arterioles in the basal ganglia and deep cerebral white matter also show thickened media but these arteries do not contain amyloid. At times, vessel walls that contain amyloid seem to be reduplicated or split. CAA predominantly affects persons older than 65 years and increases in frequency in the eighth and ninth decades.
The other type of pathology that causes small deep infarcts involves the large arteries that give rise to penetrating artery branches rather than intrinsic disease of the branches themselves. The orifices of these penetrating arterery branches could be obstructed by atherosclerotic plaque lesions. Fisher and Caplan,9 Fisher,10 and Caplan11 described the vascular pathology in these branches, and dubbed the condition intracranial branch atheromatous disease. Figure 3 contain cartoons that illustrate the location and mechanism of the pathology within the parent arteries. The orifices of the penetrating branches could be blocked by atheroma in the parent artery, atheroma could originate in the parent artery and extend into the branch (so-called junctional atheromatous plaques), or microatheromas could arise at the origin of the branch itself. Thrombus was sometimes superimposed on the atheromas and occasionally a microdissection developed in the parent artery and spread into the first millimeters of the branch.9
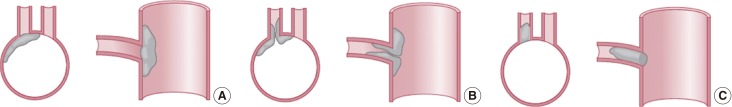
Drawing showing the arterial pathology in atheromatous branch disease: (A) plaque in parent artery obstructing a branch, (B) junctional plaque extending into the branch, (C) microatheroma formed at the orifice of a branch.
It is now possible to image intracerebral branch atheromatous disease using high resolution MRI. Plaques in the middle cerebral artery and basilar artery can be shown to impinge upon or occlude penetrating branches by MRI techniques that show axial sections of the origins of branches from the parent arteries. The location within the parent artery is critical in blocking lenticulostriate, thalamostriate, and basilar artery branches.12
Brain pathology
The major related pathology in the brain are small deep infarcts and degenerative abnormalities in the cerebral and cerebellar white matter. Lacunar infarcts are small, discrete, often irregular lesions, ranging from 1 to 15 mm in size. Inspection of these small cavities usually shows fine strands of connective tissue resembling cobwebs. The most common locations of these lacunar infarcts are the putamen and the pallidum, followed by the pons, thalamus, caudate nucleus, internal capsule, and corona radiata. Figure 4 shows a typical bold basal ganglia lacune. Rarer are lacunes in the cerebral peduncles, pyramids, and subcortical white matter. These lesions are not found in the cerebral or cerebellar cortices.

A necropsy specimen showing a cavity due to an old lacunar infarct that is located in and the medial basal ganglia (mostly the globus pallidus) and extends through the internal capsule in a patient with a pure motor hemiplegia during life.
At necropsy patients with chronic penetrating artery disease show extensive changes in white matter variously called leukoariosis and leukoencephalopathy. The combination of multiple small deep infarcts and extensive white matter abnormalities has been referred to as Binswanger disease,3,13,14 and the clinical findings are often classified as vascular dementia of the small artery type. Grossly visible in the cerebral white matter are confluent areas of soft, puckered, and granular tissue. These areas are patchy and emphasize the occipital lobes and periventricular white matter, especially anteriorly and close to the surface of the ventricles.3,13,14 Figure 5 is a myelin stained section from the cerebral hemispheres that shows a large area of myelin loss in the cerebral white matter. The cerebellar white matter is also often involved. The ventricles are enlarged, and the corpus callosum is usually small. The volume of white matter is reduced, but the cortex is generally spared. The ventricles are enlarged as a result of atrophy of the white matter. The white-matter abnormalities surrounding the ventricles may reduce the strength of the supporting tissue and allow mechanically more ventricular distension.3,13,14 The white-matter abnormalities are nearly always accompanied by some lacunes. Microscopic examination shows myelin pallor. Usually, the myelin pallor is not homogeneous, but islands of decreased myelination are surrounded by normal tissue. At times, the white-matter abnormalities are so severe that necrosis and cavitation occur. Gliosis is prominent in zones of myelin pallor.

Luxol fast blue (LFB) stained specimen of a cerebral hemisphere showing extensive loss of myelin staining.In a normal brain the entire white matter should stain uniformly blue.
Pathophysiology
Deep small infarcts are caused by decreased perfusion in the territory of penetrating arteries. Intrinsic disease of a penetrator (lipohyalinosis, CAA, CADASIL or other occlusive diseases within the course of the penetrating artery) causes a small infarct in the center of the penetrating artery supply. Figure 6 is a cartoon which illustrates this pathophysiology. Collateral circulatory capacity is very limited in the territory of these deep penetrators and occlusive changes in the adjoining penetrators can lead to infarcts that are larger than the territory of the acutely compromised penetrating vessel.

Diagram showing the relationship of a lacune in the pons to the causative penetrating artery vascular lesion. The artery beyond the region of arterial disorganization is thrombosed and thrombus has also formed in a retrograde manner extending toward the parent basilar artery (Reproduced with permission from C Miller Fisher.).
Blockage of the orifice of branch arteries by a plaque in a parent artery also causes ischemia but often the resulting infarction is larger than that resulting from intrinsic small artery disease. At times plaques can block multiple penetrators.
The pathogenesis of the extensive white matter abnormalities is controversial. Tandem ischemia in the territory of multiple penetrators and hypertensive crises with recurrent white matter edema have been posited. I favor a theory proposed by Dr. G Rosenberg and colleagues that attributes the degenerative white matter abnormalities to chronic leakage of fluid related to increased vascular permeability.15 Venous occlusive changes may contribute to the edema. Increase in vascular permeability is a very important potential mechanism of white matter abnormalities. The abnormal components of the vascular walls of penetrating arteries compromise the continence of the vessels to components of the blood that is travelling through the vessels. Matrix metalloproteins have an important role in understanding vascular permeability.15,16,17 Matrix metalloproteinase-9 levels are significantly elevated in the white matter and cerebrospinal fluid of patients with chronic white matter disease pathology.16,17 Matrix metalloproteinases disrupt the blood-brain barrier by degrading tight junction proteins found within blood vessels.17 These increased levels of metalloproteinases in thickened penetrating arteries could promote leakage of fluid from these blood vessels. Brown and colleagues found arteriolar tortuosity and described gradual thickening of the walls of periventricular veins and venules with collagen among 186 brains studied at necropsy.18 Some veins and venules were occluded and others had very narrow lumens. This chronic venous collagenosis was very extensive in some patients who had leukoaraiosis.18 Decreased venous drainage can promote brain edema. Chronic edema in perivascular areas could stimulate gliosis. Support for this theory of pathogenesis is that in CADACIL, white matter abnormalities precede brain infarcts.
Notes
The authors have no financial conflicts of interest.