Ischemic Stroke after Heart Transplantation
Article information
Abstract
Cerebrovascular complications after orthotopic heart transplantation (OHT) are more common in comparison with neurological sequelae subsequent to routine cardiac surgery. Ischemic stroke and transient ischemic attack (TIA) are more common (with an incidence of up to 13%) than intracranial hemorrhage (2.5%). Clinically, ischemic stroke is manifested by the appearance of focal neurologic deficits, although sometimes a stroke may be silent or manifests itself by the appearance of encephalopathy, reflecting a diffuse brain disorder. Ischemic stroke subtypes distribution in perioperative and postoperative period after OHT is very different from classical distribution, with different pathogenic mechanisms. Infact, ischemic stroke may be caused by less common and unusual mechanisms, linked to surgical procedures and to postoperative inflammation, peculiar to this group of patients. However, many strokes (40%) occur without a well-defined etiology (cryptogenic strokes). A silent atrial fibrillation (AF) may play a role in pathogenesis of these strokes and P wave dispersion may represent a predictor of AF. In OHT patients, P wave dispersion correlates with homocysteine plasma levels and hyperhomocysteinemia could play a role in the pathogenesis of these strokes with multiple mechanisms increasing the risk of AF. In conclusion, stroke after heart transplantation represents a complication with considerable impact not only on mortality but also on subsequent poor functional outcome.
Introduction
Stroke is a heterogeneous disease with more than 150 known causes [1], representing the fourth leading cause of death in the United States after heart disease, cancer, and chronic lower respiratory diseases [2]. After orthotopic heart transplantation (OHT), stroke represents a major cause of morbidity and mortality despite successful transplantation [3]. OHT is currently the gold standard therapy for end-stage heart failure, refractory to medical treatment [4]. Graft complications and infections are the main causes of mortality, but advancements in the fields of immunosuppression, infection prophylaxis, and surgical techniques have transformed OHT from what was once considered an experimental intervention into a routine treatment [5], with a survival rate at 5 years greater than 70 percent [6]. Neurologic sequelae after OHT are similar to the complications associated with valvular or bypass graft surgery [7], but perioperative cerebrovascular complications are more common after cardiac transplantation compared to routine cardiac surgery [8]. Ischemic stroke and transient ischemic attack (TIA) represent the most common cerebrovascular complications after cardiac transplantation, with an incidence of up to 13% [9], instead intracranial hemorrhage is an infrequent cause of stroke (2.5%) (Table 1) [7,10].

Cerebrovascular complications (ischemic and hemorrhagic stroke and transient ischemic attack [TIA]) after orthotopic heart transplantation (OHT) during perioperative-intraoperative period and during long-term follow-up
Risk factors
Stroke incidence increases with the number of preoperative stroke risk factors, although risk of stroke in the long-term was lower in recent transplant years compared with 1994 to 1997 [7]. A preoperative history of hypertension, diabetes mellitus, smoking, stroke, and markers of vascular disease are classical risk factors for stroke following cardiac surgery [7,11], but, after OHT, there are also other risk factors for ischemic stroke. Zierer et al. [12] showed that preoperative left ventricular assist device support, preoperative intraaortic balloon pump support, prolonged cardiopulmonary bypass (CPB) time, and postoperative hepatic failure are independent predictors of early neurologic complications. Older age and the presence of extracranial carotid artery stenosis over 50% seems also to increase the risk of stroke [12-14]. Furthermore, transplantation associated ischemic stroke was significantly more common in patients transplanted for dilated cardiomyopathy [15], and prior valvular disease [16], suggesting that manipulation and excision of the diseased heart may be the defining moment of intraoperative stroke. In children submitted to OHT independent risk factors for stroke include a primary diagnosis of congenital heart disease, pretransplant ventilator dependence, postoperative dialysis, infection requiring antibiotics prior to discharge, pacemaker implantation or drug treated hypertension during follow up [17]. Another risk factor for the occurrence of stroke after OHT is a previous stroke: commonly, cardiac surgery within 3 months of a stroke may carry a risk of worsening preoperative neurologic deficits [18]. Van de Beek [19] observed that cerebrovascular events tended to happen more often in patients with history of stroke and TIA at baseline; in fact, the risk of developing a stroke 5 years after OHT is greater in patients who had a prior stroke compared to patients without a history of stroke [14]. The risk of death after OHT is not increased in patients with cerebrovascular diseases [20], but patients with previous cerebrovascular disease are at an increased risk of stroke and functional decline after transplantation independent of other variables [11]. Furthermore, stroke was more likely to occur in patients with heart failure of miscellaneous cause, including valvular diseases [19].
Clinical presentation
In patients with stroke after OHT, the arterial terrItory affected determines the clinical manifestations and the type of focal neurological deficit: according to the classification of the Oxfordshire Community Stroke Project [21], Belvis [22] reported Total Anterior Cerebral Infarction in 23.1%, Partial Anterior Cerebral Infarction in 38.4%, Lacunar Cerebral Infarction in 15.4% and Posterior Cerebral Infarction in 23.1% of patients submitted to OHT. Other authors also highlight a more frequent involvement of the anterior circulation [13,23]. Clinically silent stroke detected by magnetic resonance imaging is probably more frequent [7]: not surprisingly, several of the autopsy series report the highest rates of neurologic complications with a frequency of up to 44% [24].
After OHT, an encephalopathy, reflecting a diffuse brain disorder, may occur in greater than 8 percent of adults [25]. These encephalopathic patients may be slow to emerge from anesthesia, can be agitated, and may have fluctuating impairment of cognitive and perceptual function, with occurrence of hallucinations; an improvement often occurs during the first post operative week. Some of these patients have multiple, acute, small ischemic brain lesions, suggesting multiple emboli [26]. Concomitant seizures may accompany an encephalopathy or may develop in patients with focal cortical cerebral lesion [12,19,27]. However, Malheiros [28] reported 11 transient encephalopathy with normal brain computed tomography (CT) or magnetic resonance imaging (MRI).
Intraoperative and post-operative stroke
The incidence and the pathogenesis of cerebrovascular accidents (stroke and TIA) vary according to the time of onset of symptoms after OHT; there are significant differences between perioperative stroke and long-term cerebrovascular complications.
Cerebrovascular diseases are early post-operative (less than 2 weeks) in 20% of patients and late in 80% [22]; however in the first two months after OHT, some authors reported a greater number of complications in comparison with the subsequent follow-up: 62.5% in the first 30 days [9], 80% in the first 60 days [15,27].
Table 1 shows the frequency of cerebrovascular complications during peri-intraoperative period and during long-term follow-up.
During intraoperative period ischemic stroke or TIA are mostly influenced by the underlying disease and by the surgical procedure: in these cases cerebrovascular complications may result from less common and unusual mechanisms linked to intraoperative factors, such as large fluctuation in hemodynamic parameters or embolic complications, resulting in anoxic-hypoperfusion events, causing wathershed infarcts [29]. Early after OHT, cerebrovascular complications may occur because of surgical or medical complications during postoperative course, such as mechanical support of the circulation, cardiac tamponade or arrest [7]. During early and late post-operative period, a low-grade subclinical inflammation, subsequent to surgery, may play a pathogenic role in stroke pathogenesis. Finally, during long-term follow up, in patients submitted to OHT, in addition to traditional pathogenic mechanisms, other complex mechanisms, including chronic subclinical inflammation and presence of factors enhancing thrombosis (such as homocysteine) may cause an ischemic stroke. Pathogenic mechanisms of ischemic stroke after OHT are described more fully in the following section.
Pathogenesis
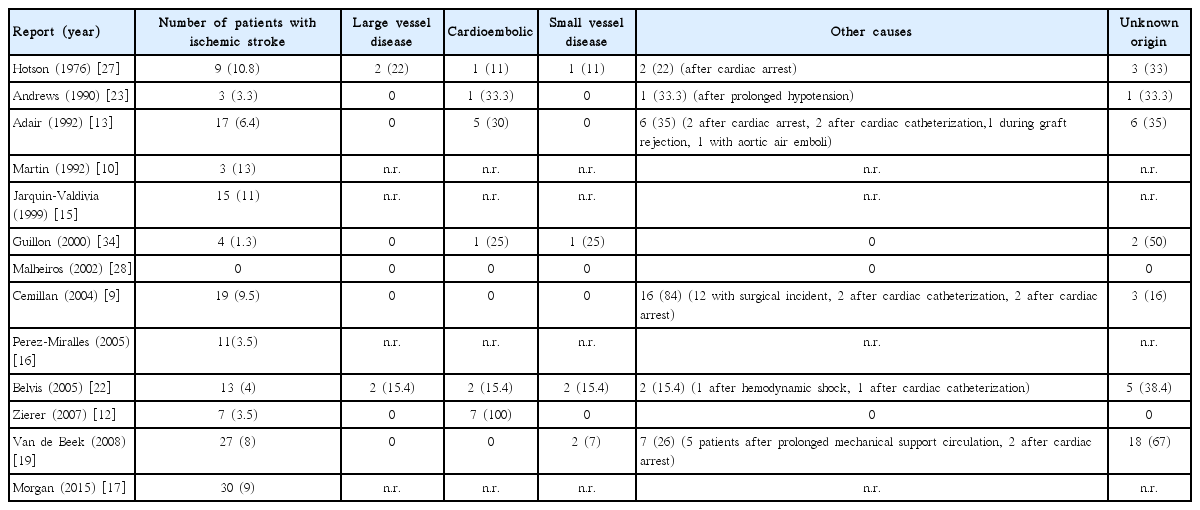
In the general population, the most common type of stroke is ischemic stroke, in comparison with hemorragic stroke that accounts for 10%-15% of all stroke cases [30]. Similarly, after OHT, ischemic stroke represents the most common cerebrovascular complication in comparison with hemorragic stroke (Table 1). According to pathogenic mechanism of ischemic stroke (TOAST classification) [31], the classical distribution of ischemic stroke subtypes in North American and European studies is the following: cardio-embolism (20%), large artery atherosclerosis (25%), small artery occlusion (25%), other unusual determined etiologies (5%), and stroke of undetermined etiology (25%) (Figure 1) [32]. Similarly, stroke subtypes may be determined according to the ASCOD criteria [33]: every patient was graded into one of the 5 predefined phenotypes (A: atherosclerosis, S: small-vessel disease, C: cardiac pathology, O: other cause, and D: dissection), assigning a degree of likelihood of causal relationship to every potential disease. According to the results of available studies providing specific information about post-OHT stroke pathogenesis (including 1764 OHT patients and 99 ischemic strokes overall, 5%; Table 2) [9,12,13,19,22,23,27,34], the ischemic stroke subtypes distribution after OHT is very different from the distribution observed in the general population with the following approximate percentages: large artery atherosclerosis 5%, cardioembolism 15%, small vessel disease 5%, unusual causes 35% and undetermined cause 40% (Figure 1).

Pathogenesis of ischemic stroke in general population (A) and in patients submitted to orthotopic heart transplantation (B).
Pathogenesis of ischemic stroke after orthotopic heart transplantation
Ischemic stroke subtypes
Large artery atherosclerosis
Extracranial large artery atherosclerosis and large artery intracranial occlusive disease represent a common pathogenetic mechanism causing an ischemic stroke [35], even after OHT [22]. A risk factor analysis in patients with stroke after OHT showed an increased risk of stroke in patients with carotid stenosis greater than 50% evaluated during preoperative ultrasonography [13]. Adult recipients of the heart transplantation have multiple factors favoring the development of extracardiac vascular diseases: the majority of the patients need to undergo multiple arterial cannulations for various reasons, with subsequent iatrogenic vascular diseases; moreover, post-transplant immunosuppressants may induce the development of vascular disease [36]. Furthermore, large artery intracranial occlusive disease also, affecting the middle cerebral artery, intracranial portion of the internal carotid artery, vertebrobasilar artery, and posterior and anterior cerebral arteries, was reported as a cause of stroke after OHT [27].
Cardioembolic stroke
In the general population the leading cause of cardioembolic stroke is atrial fibrillation (AF), especially in elderly individuals [37]. After OHT, in the immediate postoperative period, high incidence of AF has been reported [38]: 50% of postoperative AF occurs within the first two weeks. Many cardioembolic ischemic strokes and TIA, in perioperative and postoperative period, are associated with the occurrence of atrial arrhythmias (AF and atrial flutter), suggesting that these arrhythmias may play a role in the pathogenesis of these strokes [13,19,23]. Furthermore, Almassi et al. [39] reported a 5.3% incidence of stroke in transplant patients with episodes of postoperative AF compared with 2.4% in patients without postoperative AF; other authors confirmed these results [40-42]. AF occurs in approximately one-third of continuously monItored patients following cardiac surgery and is a well-known risk factor for stroke. Its initial occurrence is most common during the first 3 postoperative days, and 20% of patients have more than one episode [7]. After coronary artery bypass surgery, the evidence that the peak incidence of AF occurs on the second and third postoperative day, coinciding with the peak elevation of C-reactive protein, suggested a pathogenetic role of inflammation in the risk of development and recurrence of AF [43]; this hypothesis was confirmed by the observation that, in canine sterile pericarditis (an experimental model of postoperative AF in humans), AF was reduced by 60% after treatment with antinflammatory drugs [44]. In fact, operative trauma subsequent to cardiac surgery triggers a systemic and local inflammation response with inflammatory cytokines acting at various levels to promote structural and electrical atrial remodeling (Figure 2) [45].

Pathogenic mechanisms favoring atrial fibrillation (AF) occurrence and subsequent ischemic stroke in patients submitted to orthotopic heart transplantation (OHT). Cardiopulmonary bypass and surgical trauma can lead to the production of different proinflammatory mediators and reactive oxygen species, that determine atrial electrical and structural remodeling. Transforming growth factor-β1 is a key regulator of atrial fibrosis, modulating intercellular and cell–matrix interactions, disrupting atrial electrical conduction and predisposing to anisotropy and re-entry, fundamental substrates for AF. Another cause of atrial structural remodeling is a prolonged graft ischemia time and cardiac allograft vasculopathy. Postoperative inflammation or cellular and humoral rejection play also an important role in atrial electrical remodeling: increased levels of cytokines, modulating the function of ion channels and calcium homeostasis, induce delayed afterdepolarizations, or early afterdepolarizations. Autonomic variable reinnervation may contribute to atrial electrical remodeling, favoring AF occurrence and subsequent high risk of ischemic stroke. CPB, cardiopulmonary bypass; ROS, reactive oxygen species; TNF, tumor necrosis factor; TLR, Toll-like receptor; TGF-β1, transforming growth factor-β1; IL, interleukin; MMP, matrix metalloproteinase; PDGF, platelet derived growth factor; MPO, myeloperoxidase; ECM, extracellular matrix; Ito, transient outward potassium current; EADs, early afterdepolarizations; DADs, delayed afterdepolarizations.
Inflammation plays a particular role in atrial structural remodeling: overt inflammatory states of cardiac and noncardiac location and low-grade subclinical inflammation in postoperative states contribute to atrial fibrosis. CPB and surgical trauma can lead to the production of different proinflammatory mediators with increased levels of interleukins (IL-1β, IL-6, IL8, IL18), tumor necrosis factor, transforming growth factor-β1 and reactive oxygen species, all associated with postoperative AF [46]. High reactive oxygen species levels, generated during cardiac surgery, can modulate multiple signaling pathways and transcription factors (NF-KB, NRF2 and MAPK) leading to inflammation, apoptosis or necrosis. IL-8 can exacerbate cardiac injury; moreover IL-18 may activate pro-apoptotic signaling pathways, inducing endothelial cell death [45]. In this context, transforming growth factor-β1, activated by tumor necrosis factor, is a key regulator of fibrosis, promoting fibroblast proliferation and differentiation into collagen-secreting myofibroblasts and enhancing collagen synthesis, thereby affecting atrial remodelling [46]. Transforming growth factor-β1 produces its effects via “Smad complex” activation, regulating expression of genes involved in fibrogenesis (connective tissue growth factor and periostin) and resulting in production of the so-called matricellular protein, a profibrotic protein secreted into the extracellular matrix (ECM). Furthermore, matrix metalloproteinases activity increases in line with transforming growth factor-β1 expression within the myocardium and correlates with the level of inflammation; in particular collagen and matrix fragments produced by the action of matrix metalloproteinases 1 produce matrikines, proinflammatory and profibrotic factors [47]. The subsequent matricellular protein storage modulates intercellular and cell–matrix interactions, disrupting atrial electrical conduction and predisposing to anisotropy and re-entry, fundamental substrates for AF. The increased heterogeneity of conduction also correlates with myeloperoxidase activity and with an altered expression of atrial gap junction (connexin 40 and 43). After OHT performed with biatrial technique, atrial structural alterations originate also by surgical scars at atrial suture lines, which can also create areas of slowed conduction predisposing to formation with reentrant circuits, finally providing a substrate for AF [48]. Another cause of atrial structural alteration is a prolonged graft ischemia time predisposing to atrial arrhythmias in both, early and late postoperative periods: when myocardial preservation is not adequate, atrial ischemic damage, with subsequent endocardial fibrosis, play an important role in the pathogenesis of atrial arrhythmias [49]; cardiomyocytes exposed to ischemia–reperfusion or hypoxia–reoxygenation produce also IL-6 that favors structural remodeling. Finally, AF can occur also as a manifestation of remodeled and scarred atria associated with cardiac allograft vasculopathy [48], when ischemia involves the atrial tissue [50].
Inflammation plays also an important role in atrial electrical remodelling [51]: surgical trauma can lead to the production of different proinflammatory mediators with increased levels of cytokines, modulating the function of ion channels and calcium homeostasis. Tumor necrosis factor induces abnormal Ca2+ handling causing delayed afterdepolarizations, that represent a trigger for AF. Platelet derived growth factor may also contribute to electrical remodeling reducing action potentials duration. IL-6 is capable of modulating the response of myocardial β-adrenergic receptors and may inhibit voltage dependent Ca2+ channels [45]. Another cytokine, IL-8, can exacerbate cardiac injury by enhancing leukocyte activation and accumulation: neutrophils activation alone may be arrhythmogenic because activated neutrophils bind to cardiac myocytes causing changes in myocyte electrical activity with delayed repolarization, early afterdepolarizations, and arrest of repolarization [51]. Furthermore one of the neutrophil products that contributes to arrhythmogenicity is the inflammatory lipid mediator, platelet-activating factor that may induce AF in perioperative period, reducing potassium current and favoring early afterdepolarizations occurrence [52]. Toll-like receptors also seem to be involved in atrial remodeling in postoperative AF [51]; in particular, toll-like receptor 4 activation decreases transient outward potassium current (Ito), which increases action potential duration [53].
Many atrial arrhythmias, especially AF, have been attributed also to acute rejection [54,55]: atrial myocardial injury due to infiltration of inflammatory cells, edema, and subsequent scarring might be a consequence of cellular and humoral rejection. Repeated rejection episodes may lead to cumulative damage predisposing to atrial arrhythmias [56].
Finally, in denervated transplanted heart, over time, both sympathetic and parasympathetic reinnervation occur with a degree of reinnervation variable, incomplete and non uniform [57]: this autonomic alteration may contribute to atrial electrical remodeling, favoring AF occurrence. In fact, beta-adrenergic receptor activation, via adenylate cyclase activation, determines phosphorylation of a range of Ca2+-handling proteins (L-type Ca2+-channel, ryanodine-receptor and phospholamban) with subsequent sarcoplasmic-reticulum Ca2+ release, activating the Na+/Ca2+ exchanger and producing a depolarizing transient inward current which causes delayed afterdepolarizations [58]. Adrenergic stimulation also inhibits inward-rectifier K+-current (IK1) and enhances slow delayed-rectifier K+-current (IKs), promoting early afterdepolarizations.
Pathogenetic mechanism, causing the stroke, also influences the prognosis of such patients; in fact, post-transplantation AF was detected more frequently in patients with adverse neurologic outcome [12].
Cardioembolism may also arise by mechanisms other than AF: after standard OHT, the enlarged resultant atria may promote atrial thrombosis [59]; in this regard, Heroux observed that the incidence of echo-contrast or thrombi in the left atrium is significantly lower in patients having undergone the bicaval anastomosis technique at the time of transplantation then the standard biatrial anastomosis [14]. Despite an uneventful post-operative course, sinus rhythm and normal contractility of the heart, large thrombi could be found up to several months following transplantation [60-62]. Hotson reported a patient with an occlusion of left internal carotid artery with extensive infarction of the left cerebral hemisphere caused by a cardioembolic mechanism due to a left atrial mural thrombus [27].
Endocarditis represents a rare but life-threatening condition after heart transplantation [63]: this condition may also cause a cardioembolic stroke. Adair reported a patient with an ischemic stroke caused by an infective endocarditis on the second postoperative day, evaluated with an echocardiogram showing a mobile vegetation along the left atrial suture line [13].
Small-vessel disease
In the general population lacunar infarcts account for up to about a quarter of all ischemic strokes, constituting one of the classical stroke subtypes, and are included as a separate entity in all major ischemic stroke classification algorithms [64]. After OHT, clinical series reported lower rates of stroke caused by small vessel disease in comparison with general population [19,22,27,34]: as shown in Table 2, the incidence of small vessel disease ranges from 0% to 25% in a small series of patients (n=4) [34]; Belvis et al. [22] reported a small vessel disease in only 15.4% of patients with stroke after OHT.
Other causes
In patients submitted to OHT, ischemic stroke may be caused by less common and unusual mechanisms, linked to surgical procedures, peculiar to this group of patients. Ischemic stroke after cardiac surgery is most often the result of emboli or hypoperfusion: in fact, in approximately half of stroke patients, cerebral infarct is a watershed infarct, localized to the border zones between the terrItories of two major arteries [29].
Intraoperative factors may influence the frequency of stroke: a large fluctuation in hemodynamic parameters during surgery and the risk of intraoperative hypotension may determine an ischemic stroke. Andrews reported a right parietal infarction occurred ten days before transplantation after two episodes of cardiac arrest and prolonged systemic hypotension [23]. A generalized cerebral ischemia may be sustained by a transient fall in the mean CPB pressure [27]: Stockard [65] showed a significant correlation between CPB pressures of less than 50 mmHg and the subsequent development of neurological complications. Furthermore, patients who require CPB lasting more than 2 hours have a higher number of cerebral emboli detected by transcranial doppler ultrasound monItoring and also a higher frequency of stroke [7]. Most of these patients presented with focal neurologic signs occurring after the second day, particularly with risk factors such as mechanical support of the circulation, cardiac tamponade or arrest, suggesting that cerebral ischemic events after OHT may be attributable to postoperative cardiac dysfunction [19]. In other instances, ischemic stroke may also result from embolization of atherosclerotic debris originating in a diseased ascending aorta after cannulation or clamping [15] or from the multiple suture lines where the thrombus can grow more easily [13].
An ischemic stroke was also reported during cardiac catheterization of the right heart for endomyocardial biopsy, being a preexisting patent foramen ovale with intracardiac shunt [13].
Finally, since cardiectomy exposes the circulation to atmospheric pressure, it is possible also that small air emboli escape into arterial vessels causing a cerebral embolism [13]. A significant association between thromboembolism, stroke and the presence of a variant allele of prothrombin (G20210A) suggests also that genetic polymorphisms contribute to thromboembolic post-transplant complications [66].
Unknown causes
It is well known that many ischemic strokes (30%-40%) occur without a well-defined etiology [67]; similarly, Belvis [22] reported undetermined cause in 38.4% of patients with stroke after OHT, although other series reported that the cause is not identified up to 67% (Table 2) [19]. It has been suggested that a silent AF may play a key role in pathogenesis of some of these strokes and that P wave dispersion, an electrocardiographic index, predictor of paroxysmal AF, may represent a risk factor for cardioembolism [68]. Our previous study showed that in OHT patients, P wave dispersion is significantly higher in comparison with control subjects, correlating significantly with homocysteine plasma levels (Hcy) [69]. Hyperhomocysteinemia represents a very common finding in OHT, as a consequence of the progressive nephrotoxicity due to pharmacological therapy [70]; it could play a role in the pathogenesis of stroke in these patients with a dual mechanism: an acceleration of the progression of cardiac allograft vasculopathy and an alteration in the electrical atrial conduction increasing the risk of atrial arrhythmias in these denervated hearts.
Clinical and experimental evidences suggest that circulating Hcy is pathophysiologically linked with AF, representing a possible marker for this arrhythmias [71]. It is likely that Hcy can favor AF occurrence with multiple mechanisms: direct effects on atrial ionic channels (electrical remodeling) and biochemical damage on atrial ECM (structural remodeling) (Figure 3).

Possible role of homocysteine as a cause of ischemic stroke. Hcy can favor AF occurrence with multiple mechanisms: 1) direct effects on atrial ionic channels (electrical remodeling) (inhibition of the Ito and IKur currents, increase of IK1, increased Na+ currents) producing early afterdepolarizations and causing a focal ectopic/triggered activity; 2) biochemical damage on atrial extracellular matrix (structural remodeling) involving the activation of matrix metalloproteinases-9 and extracellular signal regulated kinase and causing subsequent atrial fibrosis with slow and a heterogeneous atrial conduction (high P wave dispersion), favoring the appearance of a vulnerable reentrant substrate. Hyperhomocysteine may also contribute to a prothrombotic state, favoring atrial thrombosis and possible subsequent ischemic embolic stroke.
Hcy may play a direct effect on potassium currents of atrial human myocytes: in particular, Hcy influences potassium channels resulting in an inhibition of the Ito (Ito channels may have functional cysteine residues that are modulated by Hcy and/or homocystine, directly causing channel inhibition) and IKur currents with a parallel increase of IK1 [72,73]. Furthermore elevated homocysteine levels significantly increased sodium currents through slowing the inactivation process and promoting the recovery process in human atrial myocytes, with resting membrane potential of myocytes more depolarized [74]. Reduced repolarizing K+ currents or increased depolarizing currents (Na+ currents) prolong action potential duration, increasing inward currents and causing membrane depolarization during phase 2 or 3 of action potential and producing early afterdepolarizations, causing a focal ectopic/triggered activity.
In addition to this electrical remodeling, Hcy produces also an atrial structural remodeling. Hcy produce a biochemical damage, affecting the structure and function of different cellular proteins also altering the redox status with relevant pathophysiological consequences (endothelial dysfunction, proliferation of vascular smooth muscle cells, alteration of the properties of interstitial fibers with remodeling of the ECM) [75]. Atrial structural remodeling is a complex process, involving the activation of matrix metalloproteinases-9 and extracellular signal regulated kinase [76]: hyperhomocysteinemia causes ECM remodeling, by the activation of extracellular signal regulated kinase-matrix metalloproteinases-9 signaling axis; furthermore among ECM proteins, collagen type I synthesis was altered by hyperhomocysteinemia [77]. The subsequent atrial fibrosis causes a slow and a heterogeneous atrial conduction, favoring the appearance of a vulnerable reentrant substrate.
High values of P wave dispersion observed in OHT patients [69] may reflect this heterogeneous atrial conduction, determined by these mechanisms, favoring the occurrence of atrial arrhytmias. Both, the vulnerable reentrant substrate and the focal ectopic/triggered activity, contributes to AF occurrence with possible subsequent cardioembolic ischemic stroke. Furthermore, hyperhomocysteinemia may also contribute to a prothrombotic state, involving an increased tissue factor expression, an attenuated anticoagulant processes, an enhanced platelet reactivity, an increased thrombin generation, an augmented Factor V activity, and an impaired fibrinolytic potential [78].
Hemorrhagic stroke
After OHT, hemorrhagic stroke is most commonly located in the deep (basal ganglia, thalamus, pons, and cerebellum) than in the cerebral lobes [9,12,13,16,19,22,23,27,34].
Pathogenesis of hemorrhagic stroke is usually caused by coagulation disorders, high pressure of the CPB and uncontrolled blood pressure [22]. Furthermore, lobar hemorrhagic stroke has been reported without these disorders in up to 5% of patients, probably due to a mechanism of hyperperfusion [13,23,27,79].
Outcome
Strokes are a significant cause of morbidity and mortality following heart transplantation [14]: cerebrovascular accidents are responsible for 7% of early postoperative deaths after cardiac transplantation [80]; furthermore, perioperative stroke is the most important neurologic complication affecting survival and it is associated with 1-year mortality [19].
Zierer [12] confirmed these data, reporting an operative mortality in patients with neurologic complications up to 15%; instead, considering the long-term mortality, five-year survival was not significantly impaired in patients with neurologic complications, though there was a trend toward high mortality in patients with neurological events (P=0.07).
After cardiac surgery, stroke prolongs length of stay in the intensive care unit and requires subsequent rehabilitation or nursing home placement: the majority of stroke patients who survive to hospital discharge are substantially disabled [81]. Longer ventilation time, longer stay in the intensive care unit, and higher incidence of pneumonia and sepsis are consistent with a complicated postoperative recovery in patients with adverse neurologic outcome [12]. The recurrence of stroke is 18% in patients having presented with a stroke following heart transplantation [14]; furthermore, patients with previous stroke before heart transplantation are also at an increased risk of stroke and functional decline after transplantation [11].
Prognosis is more favorable in cases of pediatric stroke: following a stroke, after pediatric heart transplantation, survival at 1 month, 1 year, and 5 years was 83%, 69%, and 55% respectively [17].
In conclusion, stroke after OHT represents a complication with considerable impact not only on mortality but also on subsequent poor functional outcome.
The knowledge and the recognition of the pathogenic mechanisms underlying the ischemic strokes after OHT might suggest what risk factors should be controlled in order to prevent cerebrovascular complications and to reduce post-OHT stroke occurrence.
A preoperative history of hypertension, diabetes mellitus, smoking, stroke, with markers of vascular disease are common features in patients considered at high risk prior to surgery: the presence of these risk factors suggests the need for more careful control of some further intraoperative risk factors (hemodynamic parameters during surgery, CPB time and pressure). In the late post-operative period, the hypothesis that inflammation and hyperhomocysteinemia may favor atrial remodeling with subsequent risk of silent AF occurrence, suggests that long-term EKG monItoring can be useful in unmasking paroxysmal AF episodes in selected patients with increased C reactive protein and homocysteine plasma levels, resulting in a different treatment of secondary stroke prevention.
Notes
The authors have no financial conflicts of interest.