Biomarker for Ischemic Stroke Using Metabolome: A Clinician Perspective
Article information
Abstract
Finding ischemic stroke biomarker is highly desirable because it can improve diagnosis even before a patient arrives to the hospital. Metabolome is one of new technologies that help to find biomarkers. Most metabolome-related ischemic stroke studies were done in Asia and had exploratory designs. Although failed to find specific biomarkers, they discovered several important metabolite-stroke associations which belong to three pathophysiological mechanisms: Excitotoxicity with activation of glutamate, resulting in the increase of glutamate derivatives proline and pyroglutamate; Oxidative stress with production of free radicals and perturbed concentrations of uric acid, matrix metalloproteinase-9, branch-chained amino acids, sphingolipids, homocysteine, asymmetric dimethylarginine, nitric oxide and folate cycle metabolites; and Stroke mediated inflammation, affecting phospholipid metabolism with perturbed levels of lysophosphatidylethanolamine and lysophosphatidylcholine. The discovered metabolite-stroke associations need further evaluation in prospective, high-quality studies with patients matched for age, risk factors, and medications.
Introduction
Ischemic stroke is the leading cause of disability in the United States with 6.3 million people affected by the disease [1]. Stroke affects all ethnicities, but in particular Asian countries. One study showed that compared with white populations, Chinese have a higher age-adjusted incidence of stroke, a lower mean age of stroke onset [2]. During the last 30 years, massive efforts in research led to discovery of effective stroke therapies, which are comparatively safe, but at the same time limited by time and the cost. Implementation of intravenous (IV) thrombolysis within 4.5-hour window and endovascular thrombectomy within 24-hour window from the beginning of the stroke symptoms greatly improved outcomes [3-5]; newer anticoagulants prevent more strokes from atrial fibrillation [6]. In spite all that many important questions remain unanswered and many breakthrough are still to be made. First, ischemic stroke is difficult to predict. Many people with known traditional risk factors such as hypertension, diabetes, and hyperlipidemia do not experience strokes, and others who have a healthy lifestyle may have stroke early in life without reasonable explanation. Second, quick diagnosis of stroke remains challenging in many situations when clinical presentation is vague and neuroimaging, in particular, magnetic resonance imaging is not readily available. Finally, 20% of ischemic strokes remain cryptogenic, i.e., without determined etiology. Establishing serum or urine metabolite variations associated with ischemic stroke can help solving all these questions and serve as ischemic stroke biomarkers. Discovering such biomarkers is a very difficult task which requires a good understanding of the sequence of events that leads and happens during ischemic stroke. One of the technologies which help to understand the biochemical process in the infarcted brain is metabolomics. It can be performed on different bodily tissues or fluids and identify small molecule metabolites with significantly increased or decreased values secondary to pathophysiological processes.
Metabolome technology
Metabolomics is a term used to describe measurements of multiple small molecule metabolites in biological specimens, such as urine, blood, cerebrospinal fluid and tissues. It provides a snapshot of the physiology, which can capture the unique “chemical fingerprints” that a cellular process leaves behind. Metabolomics is considered to be a recent addition to other “omics” such as genomics, transcriptomics, and proteomics. The emergence of high throughput metabolite profiling techniques has led to rapid progress from a single-metabolite association to metabolome-wide approach with increasing application in disease research including cerebrovascular disease [7]. One of the attractive features of metabolite profiling in human is a comparatively small number of human metabolites (approximately 7,000) relative to the estimated numbers of genes (25,000), transcripts (100,000) and proteins (1,000,000) [7]. These metabolites fall downstream of genetic, transcriptomic, proteomic, and environmental variation; thus, provide the most integrated and dynamic measure of phenotype and medical condition [7]. They exist in a very broad range of concentrations and exhibit remarkable chemical diversity. As such, no single instrument can measure all metabolites of the body in the single analysis. Metabolome profiling is commonly performed using nuclear magnetic resonance (NMR), where metabolites are separated by their magnetic resonance shift, or mass-spectroscopy (MS) where spectral separation is done with mass/charge ratio [7]. Both methods have advantages and disadvantages. NMR has low sensitivity, but at the same time not destructive to the sample, requires minimal preparation, quantitative, and reproducible. MS is highly sensitive, but requires sample ionization by radiation or electron beams. Resulting fragments are accelerated by electric and magnetic fields to finally display a mass spectrum showing total number of ions versus mass number. These are not recoverable, hence, destructive to the sample [8]. MS is also time consuming, and tricky to quantify [9,10]. Metabolome profiling can be “targeted” and “non-targeted.” In the non-targeted method, NMS and MS are used for simultaneous measurement of as many metabolites as possible [7,10,11]. It is mostly used for exploratory studies to compare two biological or clinical states. In targeted analysis, specifically identified metabolites are profiled. An advantage of the targeted method is its quantitative precision. A disadvantage is a limitation in the breadth of analysis, which covers several hundred metabolites in six to seven chemical classes [12]. Current MS platforms including time-of-flight, Orbitrap, and Fourier transform ion cyclotron resonance (FT-ICR) mass analyzes offer very high on mass resolution and accuracy. By coupling such MS instrumentation with high resolution chromatographic technologies such as ultra-high pressure chromatography, it is possible to resolve literally thousands of individual small molecules. Further peak analysis is being done through such databases as METLIN, KEGG, HMDB (Human Metabolome Database and others) [7,13]. Identification of peaks can also be done through the use of retention time locked libraries [11]. Combination of targeted and non-targeted approaches is applied for discovering metabolite associations. Low molecular weight small metabolites (below 1500 Da), either involved in disease, or affected by the disease process, can serve as an effective disease biomarker [14].
Metabolome profiling in acute ischemic stroke
There were many attempts to identify particular metabolites associated with ischemic stroke using rat models and human subjects. To serve as biomarker these associations should be reliable, have adequate sensitivity, specificity, and good pathophysiological explanation. Although no particular metabolite or combination of discovered metabolites yet fit these criteria, during last 10 years association of certain metabolite changes with brain ischemia were found. These metabolite changes fall into three major pathophysiologic categories: excitotoxicity or neurotoxicity [15,16], oxidative stress [17,18], and inflammation [19,20]. In this review we summarize and classify most discovered metabolite associations with ischemic stroke into mentioned above pathophysiological categories. Summary of discovered metabolite changes presented in Table 1. Below we discuss each category separately.

Summary of discovered metabolites associated with acute ischemic stroke
Excitotoxicity or neurotoxicity
Excitotoxicity was the first molecular mechanism of ischemic brain tissue injury to be identified and thoroughly studied [15]. Excitotoxicity refers to the rapid release and inhibited reuptake of excitatory amino acid (AA) and neurotransmitter glutamate as a result of energy failure. The accumulation of glutamate overstimulates plethora of downstream signaling pathways, many of which involve a surge of calcium influx, causing an increase in intracellular calcium concentration. However, different authors report contradictory findings of glutamate levels in cerebral ischemia. Increased levels of glutamate post ischemic stroke were reported by Liu et al. [21] and Schousboe [22]. They also mention that glutamine, a main precursor of glutamate, is seen in higher plasma concentrations in ischemic stroke patients [21]. Glutamine and glutamate are interconverted between astrocytes and neurons. This glutamine/glutamate cycle is essential for glutamine homeostasis and neurotransmitter glutamate generation and recycling [21,22]. Another study, though, points to decreased glutamine concentration in lacunar strokes which is explained by postischemic upregulation of glial fibrillary acidic protein expression [23]. Serum concentrations of glutamate were mentioned to be normal in the study of Ding et al. [24] It was explained by the fact that glutamate cannot easily cross the blood-brain barrier and affect plasma levels [24]. However, authors mention that glutamate derived metabolites with bidirectional flow between the blood and the brain compartments were detected in serum in altered concentrations. Proline and pyroglutamate, the important glutamate derivatives, both were found significantly decreased post-stroke [25,26]. On the other hand aromatic AA phenylalanine and its metabolite tyrosine were increased post ischemic stroke. Phenylalanine increase is thought to be a compensatory response to supraphysiological and neurotoxic quantities of glutamate, because phenylalanine depresses excitatory glutamatergic synaptic transmission via a unique set of presynaptic and postsynaptic mechanisms [21,27]. Another study associated glutamine, homocysteine, methionine, tryptophan, aspartate, alanine, and tyrosine with acute cerebral ischemia in rats with middle cerebral artery (MCA) occlusion. Authors speculate that these changes are related to excitotoxicity and oxidative stress, without providing particular detail of pathophysiology [28].
Oxidative stress
There is accumulating evidence that oxidative stress plays an important role in acute ischemic stroke (AIS) pathophysiology [29]. It presents a significant challenge to ischemic tissue and results in destruction of neurons. We summarized major changes that happen in nerve cells during acute ischemia and metabolites that released from them in Figure 1. Oxidative stress partly is a downstream consequence of excitotoxicity resulting from the rise of secondary messenger systems coupled to the enzymatic generation of free radicals. Such mechanisms may be fueled by oxygen supply which still reaches the ischemic tissue in the penumbra or after reperfusion. Increased production of superoxide anions, hydroxyl radicals, and peroxynitrite or nitrogen dioxide has been shown in infiltrating phagocytes, vascular and glial cells in the penumbra [15,17,18]. Peroxynitrite is raised as a result of the interaction between nitric oxide and superoxide. Once formed, this compound promotes lipid peroxidation, mitochondrial and DNA damage, protein nitration and oxidation, depletion of antioxidant reserves, and, as a result of multiple signaling pathways, (activation of the nuclear enzyme poly [adenosine diphosphate-ribose] polymerase) breakdown of the blood-brain barrier [20,30]. At the same time during focal hypoxic conditions, glucose metabolism changes into the anaerobic pathway. It results in increased levels of hypoxanthine, lactate, pyruvate, and uric acid. All these metabolites were observed in higher than normal serum concentrations during AIS in the studies of Liu et al. [21] and Jung et al. [23] Elevated uric acid levels that represent the final oxidation product of purine catabolism in humans, deserves special attention because it is a powerful antioxidant. Uric acid accounts for as much as two-thirds of the total antioxidant capacity of plasma [21,31,32]. High uric acid levels may be an indicator that the body is trying to protect itself from the deleterious effects of free radicals by increasing the products of endogenous antioxidants [33]. Earlier studies indicate that uric acid administration attenuates MCA wall thickening, induces passive lumen expansion, and reduced brain damage. At the same time, it did not show the ability to reduce glutamate-induced cell death and decrease area of ischemia in a rodent model by blocking generation of free radicals because uric acid could not penetrate the blood-brain barrier [34].
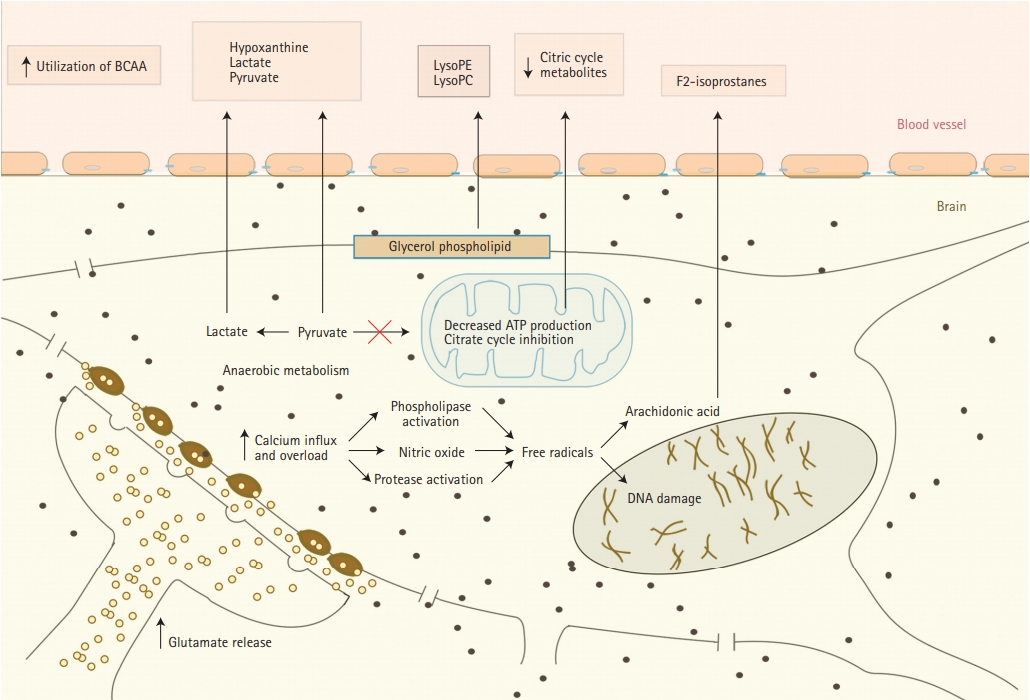
Release of metabolites from neurons into serum during cerebral ischemia. BCAA, branched-chain amino acid; LysoPE, lysophosphatidylethanolamine; LysoPC, lysophosphatidylcholine.
Free radicals peroxidation of arachidonic acid which is abundant in cerebral tissue leads to the formation of F2-isoprostanes (F2IPs). It was reported to be elevated within the first 8 hours, but not later than 24 hours after AIS, suggesting an early oxidative stress response. Matrix metalloproteinase-9 (MMP-9) is thought to be an important mediator of microvascular blood-brain barrier injury and hemorrhagic transformation after ischemic stroke. Early MMP upregulation after cerebral ischemia-reperfusion is again related to oxidative stress [35-37]. In the study of Kelly et al. [35] elevated acute plasma levels of F2IP were correlated with plasma levels of MMP-9 in AIS patients who received IV thrombolysis (tPA). This supports previously reported experimental stroke data which showed that oxidative stress may be an early stimulus for MMP activation, the bloodbrain barrier injury, or hemorrhagic transformation after AIS. Protease activity of exogenous thrombolysis may directly promote MMP activation via proteolytic cleavage of the inactive form or be related to reperfusion [35].
Another speculated consequence of AIS provoked oxidative stress is decreased adenosine triphosphate (ATP) production and inhibition of the citrate cycle. This induces the utilization of circulating branched-chain amino acids as energy compensation. Indeed, valine and isoleucine are shown to be decreased in ischemic stroke patients comparing to healthy controls [21,38]. As well, citric acid and an important intermediate in the citric cycle were observed to be reduced in stroke patients, putting more evidence that stroke is associated with inhibition of the citrate cycle [23].
Oxidative stress to vascular endothelial cell membrane, same as to neurons, plays important role in stroke pathophysiology and results in release to bloodstream of sphingolipids, as well as products of homocysteine and folic acid (Figure 2A). Sphingolipids are essential structural components of cellular membranes, playing prominent roles in signal transduction that governs cell proliferation, differentiation, and apoptosis. Sphingosine 1-phosphate (S1P) acts as a key signaling molecule and regulates lymphocyte trafficking, glial cell activation, vasoconstriction, endothelial barrier function, and neuronal death pathways which plays a critical role in numerous neurological conditions [39]. S1P consists of two molecules S1P1 and S1P2. S1P1 increases the blood brain barrier integrity (Figure 2B) and S1P2 enhances permeability (Figure 2A) [40]. Emerging data indicate multiple sphingolipid abnormalities following AIS [21,41]. Major increase of S1P levels, which again related to the oxidative stress of microendothelial cells during acute ischemia, promotes disruption of blood-brain barrier and cerebral edema [41]. Sphinganine is an intermediate in sphingolipid metabolism. It has been reported that sphingolipid activities change after stroke and correlate with stroke outcome [39].
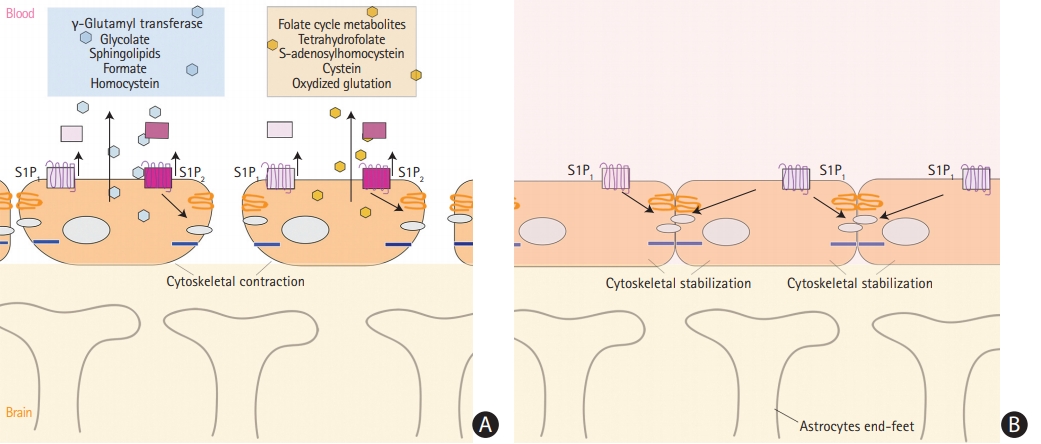
(A) Release of metabolites from vessel endothelial cells into serum during cerebral ischemia with interruption of the blood brain barrier. (B) Integrity of the blood brain barrier and no release of metabolites. S1P, sphingosine 1-phosphate.
Increased levels of homocysteine is not only a known ischemic stroke risk factor, but also an indicator and promoter of ischemic injury to endothelial cells in brain vessels [42-44]. Its detrimental effect seem to be multifactorial. Elevated total homocysteine levels induce oxidative stress to vascular endothelial cells and impair production of nitric oxide, a strong vascular relaxing factor in the endothelium [44,45]. Homocysteine enhances platelets adhesions to vessel walls, which is better described in lacunar infarctions [23]. Increased levels of homocysteine metabolites such as formate and glucolate, and decreased levels of dimethylamine, glycine, hippurate, and methanol were observed in stroke patient [23,46]. In particular a decrease in levels of dimethylamine results from impaired activity of enzyme dimethylarginine dimethylaminohydrolase [47]. In addition to this, homocysteine acts on coagulation, pro-thrombotic system [48]. In the study of Lonn et al. [49], authors attempted to lower homocysteine levels, but it failed to reduce stroke frequency. Thus, they suggested that homocysteine is just an acute phase reactant of brain ischemia and may be used as biomarker [49].
Folic acid, which remethylates homocysteine to methionine, and four other compounds of folate cycle were mentioned as biomarkers of the ischemic stroke [50]. They include tetrahydrofolate, cysteine, S-adenosylhomocystein, and oxidized glutathione [50]. Pathophysiology of cysteine and S-adenosylhomocystein elevation though may be related to comorbidities such as diabetic neuropathy. Oxidized glutathione levels could have raised during cerebral ischemia because of unbalanced production of reactive oxygen species [50].
Higher concentrations of asymmetric dimethylarginine, a circulating endogenous inhibitor of nitric oxide synthase, were associated with cerebral ischemia, but high concentrations of it are also seen with age, smoking, diabetes, hypertension, and homocysteinemia [51,52]. In the rat AIS model and dynamic metabolite evaluation, two main compounds were associated with AIS. Level of malonic acid was increased at 3 hours and peaked at 12 hours after reperfusion. Level of glycine has peaked at 6 hours, declined at 12 hours after reperfusion and finally dropped to the baseline [53].
Gamma-glutamyltransferase (GGT) is the enzyme responsible for the extracellular catabolism of glutathione, the main thiol intracellular antioxidant agent in mammalian cells [54,55]. Although mostly abundant in liver and commonly related to alcohol consumption, GGT was associated with increased risk of ischemic stroke and intracerebral hemorrhage in Korean population [56]. Besides that, GGT has been associated with mortality from cardiovascular disease and poor outcomes after ischemic stroke [55,57]. In the study of D’Ambrosio et al. [55] increased levels of GGT correlated with higher National Institutes of Health Stroke Scale (NIHSS), suggesting larger ischemic infarctions. GGT is abundantly present in endothelial cells of brain vessels, and serve as a biomarker of blood-brain barrier disruption [55]. When oxidative stress and inflammation affect ischemic brain, dynamic changes in GGT activity reflect evolving changes of endothelial function toward endothelial dysfunction [58]. This way GGT may serve as a candidate biomarker of early cerebral ischemia.
Inflammation
The immune-mediated inflammatory response after AIS is led by number of damage associated molecules, including chromatin-associated protein, heat shock protein, ATP, S100 proteins, heparin sulfate, DNA, and RNA. The first immune cells to respond are intrinsic brain microglia and neutrophils that exacerbate oxidative stress and blood-brain barrier damage [59,60]. Further, after activation and release of mediators, glial cells attract more neutrophils, monocytes, and lymphocytes [61]. Ischemia and reperfusion also trigger activation of the complement system [62]. Monocytes, monocyte-derived macrophages, dendritic cells, natural killer cells, and lymphocytes regulate postischemic inflammation, and specific populations of primed or unprimed T-cells might have beneficial or detrimental roles [63,64].
Stroke-induced inflammatory response involves the increase in several proinflammatory cytokines which activate the indoleamine-2,3-dioxygenase enzyme [65]. The increased expression of this enzyme leads to a decrease of AA tryptophan and the production of kynurenine [21]. A metabolite of tryptophan 3-indole propionic acid, produced by gut microflora in mammals was found to be decreased in serum of stroke patients [66].
Ischemic stroke inflammatory response affects phospholipid metabolism [21,67]. In particular, inflammation is suggested as a cause of perturbed metabolism of phosphatidylethanolamine (PE), phosphatidylcholine (PC), and lysophosphatidylcholine (LysoPC) including LysoPC (14:0), LysoPC (18:2), LysoPC (16:0), LysoPC (18:1), LysoPC (18:0) and LysoPC (20:1). All these are important signaling molecules with diverse biological function and are intermediates of glycerophospholipid metabolism, which is a backbone of neural membrane (Figure 1) [21,68,69]. These molecules had decreased levels in chronic ischemic stroke patients, as well as patients with Alzheimer’s disease and mild cognitive impairment, but may be elevated in AIS [70]. Another two metabolites associated with abnormal phospholipid metabolism in ischemic stroke are lysophosphatidylethanolamine (LysoPE) and phosphatidylserine (PS) [68]. LysoPE is a hydrolysis product of PE by phospholipase A2, which plays a role in cell-mediated cell signaling and activation of other enzymes. PS is a glycerophospholipid in which a phosphorylserine moiety occupies a glycerol substitution site [68,71,72].
One study evaluated role of metabolome in the prediction of AIS occurrence. Low concentrations of specific LysoPC (16:0) were significantly associated with stroke recurrence. LysoPC (20:40) arose as a stroke recurrence biomarker in combination, increasing the predictive value of the ABCD score. LysoPC (22:6) was described as a potential biomarker for large artery atherosclerosis [73].
Change in bio-energy homeostasis and other disturbed metabolites
Oxidation of fatty acids, which can pass the blood-brain barrier, accounts for up to 20% of the total brain energy [74]. Several studies suggested excess consumption of fatty acids in post-stroke patients. It is initiated by the protective regulation of central nervous system to restore the bio-energy homeostasis in the face of the energy failure induced by stroke. In this view, altered fatty acid profile, in particular, a significant decrease, their plasma level compared to healthy individuals was observed in several studies [24,75,76]. In particular, there were decreased plasma levels of hydroxyoctadecadienoic and hydroxyeicosatetraenoic acids [50]. In addition, hydroxyoctadecadienoic acid was associated with atherosclerosis, and hydroxyeicosatetraenoic acid reported to be a biomarker of vasoconstriction in cerebral and mesenteric arteries [77,78].
Carnitine is another metabolite observed in AIS and associated with its poor clinical outcomes. Carnitine is thought to be released from damaged muscle cells in other conditions, though the source of its increase in AIS is not well explained. Higher levels of N-acetylneuraminic acid and creatinine were observed in stroke patients. Emerging data indicate that both these components are related to acute cerebral ischemia, but no good explanation of pathophysiology of their elevation is provided [79,80]. Another study performed analysis of targeted metabolome data of 105 serum metabolites, including acylcarnitines, AAs, phospholipids, and hexone. It did not reveal any association of these metabolites with ischemic stroke, but showed an association with myocardial infarction [81].
Identifying patients at risk for ischemic stroke is another direction of metabolome research. One study separated nine metabolites associated with risk of ischemic stroke: low levels of N6-acetyl-l-lysine, 5-aminopentanoate, cadaverine, 2-oxoglutarate, nicotinamide, l-valine, S-(2-methylpropionyl)-dihydrolipoamide-E and ubiquinone, and elevated levels of homocysteine sulfonic acid. Further analysis showed that these metabolite biomarkers are specifically related to stroke occurrence, and unrelated to other factors such as diabetes or smoking. Lower levels of lysine catabolites in thrombotic stroke risk patients, as compared to the controls, supports targeting these compounds to investigate as novel biomarkers for early and non-invasive detection of a thrombotic stroke [82].
Discussion and future perspectives
Metabolome profiling is comparatively new technology for evaluation of pathophysiology of different medical conditions. It has been applied in stroke for more than ten years with several studies being done on animals (rodents) and patients. The vast majority of these studies used untargeted metabolome profiling and had an exploratory design. They compared metabolome profile in patients with AIS and controls without ischemic stroke in hopes of finding a difference between the two. Later they try to explain this difference using knowledge from known pathophysiologic pathways of cerebral ischemia. With few exceptions, results of these studies are either inconsistent or non-reproducible. Therefore, conceivably the era of metabolomics in stroke and specifically in AIS is infancy stage and major advances yet to be made. Despite the fact the most of the studies targeted to find a distinguished chemical or biomarker for ischemic stroke, they could have been more beneficial to better characterize biochemical process happening in ischemic brain tissue or finding particular chemical reactions which could be aborted in order to prevent the development of stroke. Apparently, due to incomplete understanding of the underlying molecular biology, the pathophysiology of ischemic stroke is not yet understood well to explain all discovered metabolite changes.
Most of the metabolites discovered in these studies fall into three stroke pathophysiological categories, including excitotoxicity with glutamate, oxidative stress with the formation of free radicals, and cell-mediated inflammatory response. Many of these studies show either similar metabolites or different metabolites of the same pathophysiologic pathway, which could be explained by the difference in the methods of evaluation such as NMR or MS. Interestingly, this finding was not noticed yet in the literature. All this gives optimism for the future of metabolomics in understanding/discovering early biomarkers of AIS, which would determine future directions in stroke management and therapeutics. On the other hand, we understand that science is still far away from finding a reliable stroke biomarker.
All these studies better characterize pathophysiology of cerebral ischemia and besides discovering a biomarker may also find possible applications of new drug therapies, in particular, neuroprotective agents. For example, reducing blood glutamate concentrations with plasma glutamate dialysis or reducing the damage effect of glutamate excitotoxicity by modulating downstream signaling pathways of glutamate, such as calcium influx [20]. A better understanding of oxidative stress can help to find new applications of antioxidant therapy and explain their previous failures such as uric acid, which showed synergic neuroprotection in administration with alteplase, blocked the formation of free radicals in the vessel wall, but did not cross the blood-brain barrier [34,83]. Developing a medication which can reduce inflammation in AIS should help to understand how to “freeze” core/penumbra ratio and potentially extend IV tPA or endovascular thrombectomy window for AIS patients. Pilot studies indicate that fingolimod, commonly used in multiple sclerosis, can act on sphingosine-1-receptor, limits the inflammation, migration of lymphocytes to the brain, and inhibits local activation of microglia and macrophages [20]. If administered along with alteplase, it results in lower circulating lymphocytes, smaller lesion volumes, less hemorrhage, and attenuated neurological deficits according to the NIHSS, compared with alteplase alone [20,84]. As such, metabolomics in the future should discover many other yet unknown pathophysiological pathways and targets for drug application.
The use of metabonomics as a research tool carries limitations common to diagnostic studies and systems biology studies, as well as those unique to metabolic profiling. In most mentioned above studies, patients’ groups were not matched for vital characteristics and did not have stringent exclusion criteria. Stroke risk factors such as diabetes, hypertension, dyslipidemia, and arterial disease confer distinct metabolic profiles; it is not known whether groups matched for such characteristics would retain implied significant metabolic distinctions, particularly given the limited number of participants, as a small number of observations (samples) produce a large number of variables (metabolites). This results in an increased likelihood of type I error. Use of such medications as statins was not considered by investigators. Statins may both produce signals in the plasma/serum and also induce a physiological response that may be detected in the plasma metabolome [85]. Although some studies elaborated on the timing of bodily fluid collection in relation to acute stroke, none of them accounted for the size of ischemic stroke and its possible effect on metabolome.
Conclusions
Metabonomics offers a general overview of the physiological status of an individual and gives an effective platform to study a complex disorder with multifactorial etiology. Future metabolome stroke studies will continue investigating already established as well as new, yet unknown, pathways, and pathophysiologic mechanisms. It should be driven by an ultimate desire to understand brain ischemia to the point which is sufficient for the development of a stroke biomarker and a neuroprotective agent which may not stop, but at least slowdown conversion of penumbra to ischemic core. As for now the majority of the studies enrolled small numbers of comparatively diverse patients (small and large vessel disease within an extended time interval). We expect to see more clinical trials with a larger number of patients, which should take into account such parameters as ischemic stroke size, vascular distribution, and time from stroke onset. Discovering unique metabolic characteristics of ischemic strokes in hyperacute vs. acute vs. chronic stage, or large vs. small vessel disease is of greatest importance to clinician to make an impact on stroke care: diagnosing a type of stroke by medics in the ambulance and triaging to the hospital which can provide appropriate level of care.
Therefore, more prospectively followed high-quality clinical trials with carefully selected patients matched for risk factors, stratified for stroke etiology are required for accurate biomarker definition and to improve understanding of the pathological processes in ischemic stroke.
Notes
Disclosure
The authors have no financial conflicts of interest.
Acknowledgements
Authors express acknowledgement to Claire Delpirounouh for helping with figures.