Hypertriglyceridemia: A Neglected Risk Factor for Ischemic Stroke?
Article information

Abstract
Hypertriglyceridemia is caused by defects in triglyceride metabolism and generally manifests as abnormally high plasma triglyceride levels. Although the role of hypertriglyceridemia may not draw as much attention as that of plasma cholesterol in stroke, plasma triglycerides, especially nonfasting triglycerides, are thought to be correlated with the risk of ischemic stroke. Hypertriglyceridemia may increase the risk of ischemic stroke by promoting atherosclerosis and thrombosis and increasing blood viscosity. Moreover, hypertriglyceridemia may have some protective effects in patients who have already suffered a stroke via unclear mechanisms. Therefore, further studies are needed to elucidate the role of hypertriglyceridemia in the development and prognosis of ischemic stroke.
Introduction
Stroke is a highly prevalent disease, especially in developing countries [1,2]. According to the Guidelines for the Primary Prevention of Stroke published by the American Heart Association/American Stroke Association (AHA/ASA), the risk factors for stroke include dyslipidemia, overweight and obesity, metabolic syndrome, etc [3,4]. Hypercholesterolemia is highly prevalent in ischemic stroke patients, and approximately 45% to 60% of them are simultaneously affected by it, according to large clinical trials [5,6]. Although the relationship between hypercholesterolemia and ischemic stroke remains uncertain [7-9], the success of statins in the prevention of ischemic stroke confirms the important role of cholesterol, especially low-density lipoprotein cholesterol (LDL-C), in clinical practice [10-12]. Hypertriglyceridemia, another type of dyslipidemia, has received lesser attention than hypercholesterolemia, as its role in ischemic stroke remains controversial [7,13,14]. However, an increasing number of studies have found a close relationship between plasma triglycerides (TGs) and the development, duration, and prognosis of ischemic stroke. In this paper, we reviewed the pathophysiology of TG metabolism, the association between hypertriglyceridemia and ischemic stroke, the underlying mechanisms, and its clinical significance.
Triglyceride metabolism and hypertriglyceridemia
Plasma TG levels are regulated by the balance of synthesis, lipoprotein lipase (LPL)-mediated hydrolysis, and hepatic remnant clearance (Figure 1) [15]. Plasma TGs mainly exist within triglyceride-rich lipoproteins (TRLs), such as chylomicron (CM), very low-density lipoprotein (VLDL), and their remnant particles. Nascent CMs synthesized by enterocytes have a higher TG-to-cholesterol mass ratio and primarily consist of apolipoprotein (Apo) B-48 and A-1 [16]. These particles range from 75 to 3,000 nm in diameter, depending on the dietary lipid mass [17]. After interaction with high-density lipoprotein (HDL), the nascent CM becomes mature and acquires ApoC and ApoE on its surface. In tissue capillaries, these CMs bind to LPL, which is activated by ApoC2, thereby inducing lipolysis in TGs. Alternatively, VLDL is constantly synthesized and secreted by the liver and primarily contains TGs with ApoB-100, ApoC, and ApoE on the surface. Nascent VLDL also requires interaction with HDL to become mature and transform into smaller particles as VLDL remnants after lipolysis by LPL. The liver is a major organ that removes remnant particles from the blood, predominantly through mechanisms mediated by receptors, including ApoE, the LDL receptor (LDLR) family, and cell-surface heparin sulfate proteoglycans on hepatocytes [18,19].
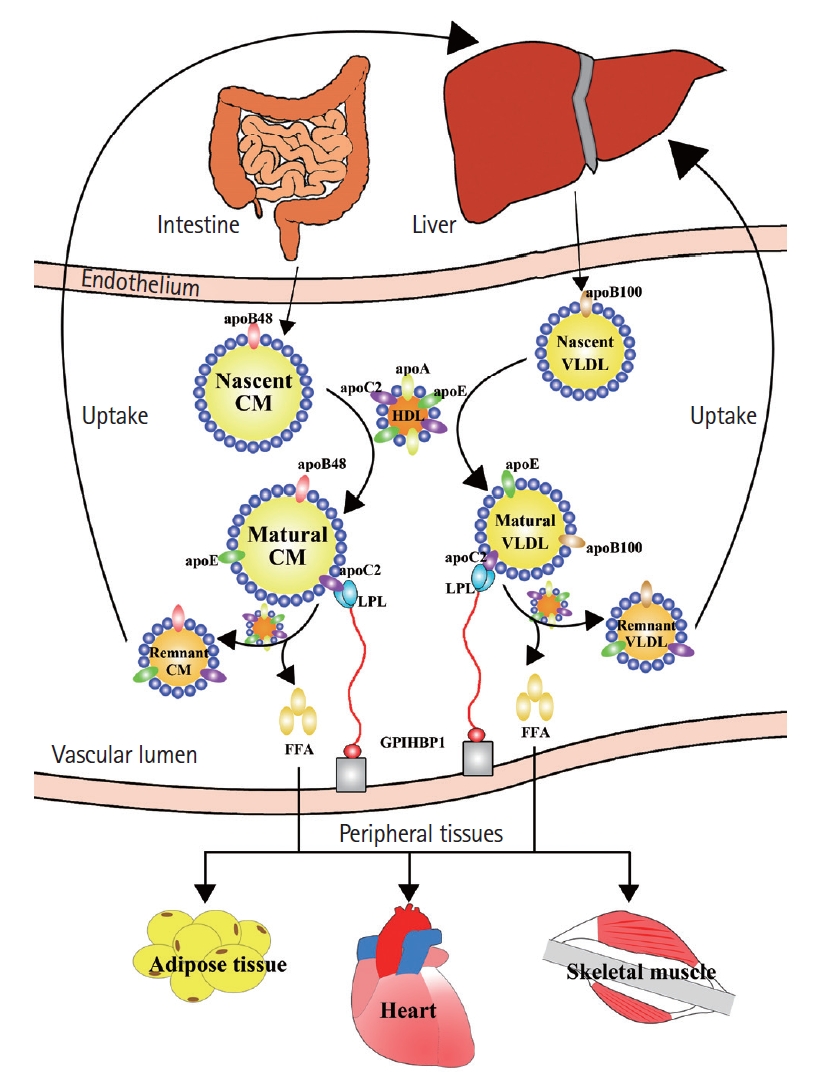
Metabolism of triglycerides [15]. Triglycerides are incorporated into chylomicron (CM) by enterocytes and into very low-density lipoprotein (VLDL) by hepatocytes. Nascent CM has apolipoprotein B48 (apoB48) on its surface, while nascent VLDL has apolipoprotein B100 (apoB100). After interaction with high-density lipoprotein (HDL), these two types of lipoproteins become mature and can be hydrolyzed by lipoprotein lipase (LPL) anchoring to the vascular endothelium. As a result, free fatty acids (FFAs) are released from CM and VLDL and utilized by peripheral tissues, such as adipose tissue, heart, and skeletal muscle. Mature CM and VLDL become remnant particles after continuous interactions with LPL and HDL and are finally taken up by the liver. GPIHBP1, glycosylphosphatidylinositol anchored high-density lipoprotein binding protein 1.
Hypertriglyceridemia is characterized by abnormally high blood TG levels. According to the Third Report of the National Cholesterol Education Program–Expert Panel on Detection, Evaluation, and Treatment of High Blood Cholesterol in Adults (NCEP-ATPIII), the concentrations of fasting TGs are stratified into four levels: normal, borderline high, high, and very high (Table 1) [20]. Any factor that interrupts the metabolic process of TGs can result in hypertriglyceridemia [15,21]. Borderline high TGs (1.70 to 2.25 mmol/L) are mostly derived from acquired factors, such as overweight and obesity, physical inactivity, cigarette smoking, excess alcohol intake, and high carbohydrate intake. High values of TGs (2.26 to 5.64 mmol/L) generally result from a combination of acquired and genetic factors. Several individuals in this classification have insulin resistance and metabolic syndrome. Family clustering was also observed in three patterns: familial combined hyperlipidemia, hypertriglyceridemia, and dysbetalipoproteinemia. Very high TGs (≥5.65 mmol/L) are typically denoted as catabolic defects of TRL, also attributed to both acquired and genetic factors. However, genetic factors may play a more dominant role because overt genetic alterations are frequently observed in this classification. Genetic defects in LPL, glycosyl-phosphatidyl-inositol anchored high-density lipoprotein binding protein 1 (GPIHBP1) or ApoC2, and the excessive expression of ApoC3, angiopoietin-related protein (Angptl) 3, and Angptl4 are known to induce severe hypertriglyceridemia [15,21].

Triglycerides and ischemic stroke
Effects of triglycerides on the risk of ischemic stroke
The relationship between TGs and ischemic stroke has been widely studied in the recent decades (Table 2) [8,13,22,23]. Ample evidence has shown that hypertriglyceridemia is a risk factor for ischemic stroke, although this evidence has not been confirmed. In a prospective cohort study covering 267,500 Chinese individuals with an average follow-up period of 6 to 19 years, investigators found that for every 1 mmol/L increase in TG levels, the multifactorial adjusted hazard ratio (HR) and 95% confidence interval (CI) for ischemic stroke was 1.07 (95% CI, 1.05 to 1.09) [24]. In a meta-analysis of risk factors for ischemic stroke in Asians, the researchers evaluated 2,611 patients diagnosed with ischemic stroke and 2,833 controls. Using a random-effects model, hypertriglyceridemia was observed to be significantly associated with ischemic stroke (P=0.007) [25]. In a prospective cohort study involving 42,005 volunteers aged 20 to 80 years with an average follow-up period of 3.6 years, the researchers found that after adjusting for age, body mass index (BMI), systolic blood pressure (SBP), diastolic blood pressure, cigarette smoking, alcohol consumption, use of lipid-lowering drugs, diabetes, history of hypertension, family history of cardiovascular disease, and other confounding factors, whether in men or women, high TG levels were associated with an increased risk of ischemic stroke (men: adjusted HR, 1.06; 95% CI, 1.00 to 1.12 and women: adjusted HR, 1.12; 95% CI, 1.01 to 1.23); additionally, this association was more notable among women [26]. In another nested case-control study, 5,475 ischemic stroke cases, 4,776 intracerebral hemorrhage cases, and 6,290 common control subjects without prior history of cardiovascular disease, cancer, or lipid-lowering, anticoagulant, or antiplatelet treatment at baseline were enrolled. The results demonstrated that plasma TG concentrations were weakly correlated with ischemic stroke risk (relative risk [RR], 1.02; 95% CI, 1.00 to 1.04 per 30% higher TGs) [8]. In a retrospective study, researchers used the Optum Research Database; additionally, through multiple screening and multivariate analyses, they showed that patients with hypertriglyceridemia had a significantly increased risk of nonfatal stroke (HR, 1.27; 95% CI, 1.14 to 1.42; P<0.001) [27].
Effects of triglycerides on the risk of ischemic stroke
In patients with diabetes, lower TGs seem to be protective factors for stroke. In one study, using Tukey’s multiple comparison, researchers found that higher TG levels were significantly associated with an increased risk of small-vessel occlusion, a subtype of ischemic stroke, in diabetes mellitus patients (P=0.013) [28]. In a community-based, prospective cohort of American Indians (median follow-up 17.7 years), researchers studied 3,216 participants (40% men, 41% with diabetes), who had no cardiovascular disease at baseline. In patients with diabetes, hypertriglyceridemia plus low HDL-C levels were associated with a 2.13-fold greater HR and 95% CI (1.06 to 4.29) for stroke (P=0.060) when compared with those in individuals with normal blood glucose [23]. In diabetic patients with LDL-C controlled by statins, the researchers divided the patients into a high TG group (2.26 to 5.54 mmol/L, n=5,542) and a normal TG group (<1.70 mmol/L, n=22,411). After adjusting for age, sex, race/ethnicity, smoking status, blood pressure, hemoglobin A1C, serum creatinine, presence of ischemic heart disease, study site, and other influencing factors, a multivariate analysis showed that the high TG group (2.26 to 5.54 mmol/L) had an increased nonfatal stroke incidence—by 23% (RR, 1.23; 95% CI, 1.01 to 1.49; P=0.037)—when compared with the normal TG group [29]. In a cross-sectional study of 223,612 type 2 diabetes mellitus patients, a multivariate analysis showed that low TGs (<1.70 mmol/L) were associated with a reduced risk of nonfatal stroke in patients who suffered from diabetes for no more than 15 years (odds ratio [OR], 0.95; 95% CI, 0.90 to 1.00;P=0.0504). However, in individuals with diabetes aged ≥15 years, low TG levels were significantly associated with an increased risk of nonfatal stroke (OR, 1.58; 95% CI, 1.45 to 1.72;P<0.001) [30].
Moreover, hypertriglyceridemia is a risk factor for stroke irrespective of age. In a cross-sectional study on acute ischemic stroke (AIS) in children, hypertriglyceridemia was more common in children with arterial ischemic stroke than in those without stroke [31]. In a national cohort study of 5,680,055 participants aged 20 to 39 years who had not taken statins, the results revealed that increased TG levels were associated with a higher risk of stroke (P<0.001) [32]. After adjusting for common risk factors, TG levels were still an independent predictor of cardiovascular events (death, myocardial infarction, and stroke) (adjusted HR, 1.20; 95% CI, 1.14 to 1.26;P<0.001) [32]. In a retrospective study, Huang et al. [33] explored the relationship between TGs and the first ischemic stroke in elderly hypertensive patients in a Chinese community. From January 2010 to December 2011, 3,249 elderly hypertensive patients were recruited, including 1,455 men and 1,794 women, with an average age of 71.36±7.18 years and an average follow-up period of 5.5 years. A multivariate Cox regression analysis revealed that elevated TG levels were significantly associated with an increased risk of first ischemic stroke (P=0.002 for trend) after adjusting for various confounding factors, such as sex, age, BMI, SBP, and fasting blood glucose [33].
In certain cases, hypertriglyceridemia is a significant risk factor for stroke. In a prospective study conducted in a rural population in southern China, 4,081 participants aged 35 to 75 years with no history of stroke had an average of 5.6 years of follow-up, and the results revealed that the hypertriglyceridemic waist phenotype (a waist circumference [WC] ≥90 cm and TG level ≥2.00 mmol/L for men or a WC ≥85 cm and TG levels ≥1.50 mmol/L for women) was significantly associated with an increased risk of ischemic stroke (HR, 1.71; 95% CI, 1.05 to 2.78), after adjusting for confounding factors (i.e., age, sex, smoking status, drinking status, history of diabetes mellitus, history of hypertension, total cholesterol level, HDL-C level, and LDL-C level) [34]. In an Atherosclerosis Risk In Communities study, the fasting plasma remnant-like particle cholesterol (RLP-C) and TGs in low-density lipoprotein (LDL-TG) levels were measured in 9,334 participants without common cardiovascular diseases, and were followed up for 16 years to observe the incidence of cardiovascular events (coronary heart disease and ischemic stroke). The LDL-TG levels were correlated with TG levels (r=0.64, P<0.0001); additionally, LDL-TG was significantly associated with a higher risk of ischemic stroke after adjusting for age, sex, race, total cholesterol, HDL-C, SBP, antihypertensive medication use, current smoking, and diabetic status in both categorical and continuous analyses (P<0.0001 and HR, 1.47; 95% CI, 1.13 to 1.92;P<0.005, respectively) [35]. Similarly, in a nested case-control study, researchers recruited 912 myocardial infarction, 1,146 ischemic stroke, 1,138 intracerebral hemorrhage cases, and 1,466 common control subjects. Nuclear magnetic resonance spectroscopy was used to measure 225 metabolic markers in baseline plasma samples. They found that TGs within most VLDL, small LDL, and small and medium HDL were positively associated with ischemic stroke. For intermediate-density lipoprotein (IDL), medium and large LDL, and very large HDL, no obvious association was observed. However, the TG concentrations were negatively correlated with ischemic stroke in large HDL (OR, 0.89; 95% CI, 0.81 to 0.98) [14]. In a study evaluating the correlation between Polish AIS and metabolic syndrome, researchers included 672 patients (387 women and 285 men) aged 32 to 93 years. The stratified analysis showed that after adjusting for age, sex, and BMI, there was a significant association between hypertriglyceridemia and stroke in patients with metabolic syndrome (adjusted OR, 4.35; 95% CI, 2.78 to 9.43;P<0.001) [22]. Moreover, genetic variants that promoted hypertriglyceridemia, such as the LPL S447X, LPL HindIII, and Apo A5 gene promoter region T-1131C polymorphisms, were associated with an increased risk of ischemic stroke [36-38].
However, in patients receiving statin therapy, TG may not be a risk factor for stroke. In a longitudinal observational cohort study that evaluated the risk of hypertriglyceridemia and cardiovascular disease in patients receiving conventional statin control of LDL-C, after adjusting for age, sex, race/ethnicity, BMI, smoking status, blood pressure, diabetes, use of insulin, history of myocardial infarction, stroke or other ischemic heart disease, serum creatinine, study site, and other factors, a multivariate analysis showed that the incidence of nonfatal strokes in the high TG group was slightly higher than that in the normal TG group; however, there was no significant difference (HR, 1.09; 95% CI, 0.89 to 1.33; P=0.42) [39]. In a retrospective study that explored the correlation between elevated TGs and an increased cardiovascular risk, the researchers found that among individuals over 45 years of age taking statins, compared with the pre-matched comparator cohort (TG levels less than 1.70 mmol/L and HDL-C levels greater than 0.45 mmol/L) (n=32,506 patients), the risk of nonfatal strokes was significantly increased in the elevated TG cohort (≥1.70 mmol/L) (n=27,471 patients) (HR, 1.14; 95% CI, 1.04 to 1.24; P=0.004). However, after removing HDL-C and other factors in the multivariate analysis model, no significant association was observed between TG levels and nonfatal strokes (HR, 1.08; 95% CI, 0.98 to 1.19; P=0.12) [40].
However, contradictory results exist regarding the relationship between TGs and ischemic strokes. In a prospective cohort study, 51,620 volunteers without a history of myocardial infarction, stroke, or cancer, who had undergone three health examinations between 2006 and 2010 were followed up for incident stroke occurrence. After adjusting for sex, age, mean TG level, use of lipid-lowering agents, education, income, smoking status, drinking status, physical activity, diabetes, hypertension, BMI, estimated glomerular filtration rate, and high-sensitivity C-reactive protein (CRP), multivariate analyses showed no significant association between TGs and ischemic stroke events (per standard deviation [SD] increase 0.99; 95% CI, 0.92 to 1.06; P=0.129) [41]. The sensitivity analyses led to the same conclusion (per SD increase 1.00; 95% CI, 0.93 to 1.06; P=0.181) [41]. In a population-based cohort study investigating early-onset ischemic strokes, researchers included 961 patients with a first-ever ischemic stroke between 25 and 49 years of age and 1,403 frequency-matched stroke-free controls. A multivariate logistic regression analysis found that there was no significant association between high TGs and ischemic stroke (adjusted OR, 1.19; 95% CI, 0.94 to 1.53) [42].
Notably, a nonfasting TG level, rather than a fasting TG level, has been attracting more attention as a strong predictor of ischemic stroke. In a prospective study of 26,509 initially healthy women in the United States (20,118 fasting and 6,391 nonfasting) with a follow-up of a median of 11.4 years, nonfasting TGs, rather than fasting TGs, maintained a strong independent relationship with cardiovascular events, including ischemic stroke, in fully adjusted models (fully adjusted HR, 4.48; 95% CI, 1.98 to 10.15; P<0.001 for trend) [43]. One of the most remarkable studies on the relationship between nonfasting TGs and ischemic stroke is a prospective, population-based cohort study based on the Copenhagen City Heart Study (CCHS). 44 This study included 13,956 participants, wherein the TG levels ranged from normal (<1.00 mmol/L) to very high (≥5.00 mmol/L), and had a follow-up of up to 31 years, during which 1,529 participants developed ischemic stroke. After a multivariate adjustment for the total cholesterol level, alcohol consumption, smoking, hypertension, atrial fibrillation, and lipid-lowering therapy, with further adjustment in women for postmenopausal status and hormone therapy, there was a 15% increased risk (95% CI, 0.09 to 0.22) of ischemic stroke for each 1.00 mmol/L increase in nonfasting TGs. After multivariate adjustment, the HRs for ischemic stroke among men and women with the highest nonfasting TGs (5.00 mmol/L or greater) compared with those with the lowest level (1.00 mmol/L) were 2.5 (95% CI, 1.30 to 4.80) and 3.8 (95% CI, 1.30 to 11), respectively. As nonfasting TGs increased from <1.00 to >5.00 mmol/L, the incidence of ischemic stroke increased from 2.6% to 5.5% in men under 55 years old and from 1.9% to 4.0% in women. Among men and women over the age of 55 years, the incidence increased from 8.1% and 5.8% to 16.7% and 12.2%, respectively. In the cross-sectional study included in the CCHS, men with a previous ischemic stroke versus controls had nonfasting TG levels of 2.16 mmol/L (interquartile range [IQR], 1.48–2.93) versus 1.67 mmol/L (IQR, 1.18 to 2.42) (P<0.01); the corresponding values for women were 1.89 mmol/L (IQR, 1.37 to 2.59) versus 1.44 mmol/L (IQR, 1.02 to 2.05) (P/i<><0.05) [44]. Similar results were reported in 2011 when the CCHS reached a 33-year follow-up [45]. Moreover, increasing cholesterol levels were not associated with the risk of ischemic stroke except in men with cholesterol levels >9.00 mmol/L versus <5.00 mmol/L, with a HR of 4.40 (95% CI, 1.90 to 10.60) [45]. In the Circulatory Risk in Communities Study, the researchers found that the nonfasting TG levels rather than the fasting TG levels could predict the risk of ischemic stroke in women (multivariable HR, 1.39; 95% CI per 1 mmol/L increment, 1.24 to 1.57; P for trend=0.013 vs. multivariable HR, 0.90; 95% CI per 1 mmol/L increment, 0.68 to 1.19; P for trend=0.887, respectively), while in men, neither could predict the risk of ischemic stroke (multivariable HR, 1.04; 95% CI per 1 mmol/L increment, 0.94 to 1.15; P for trend=0.219 vs. multivariable HR, 1.10; 95% CI per 1 mmol/L increment, 0.94 to 1.30; P for trend=0.074, respectively) [46]. Similarly, researchers found that nonfasting TGs could replace fasting TGs as a risk predictor of future cardiovascular events, including myocardial infarction, stroke, or cardiac death in Japanese diabetic patients [47].
Effects of triglycerides on the severity and prognosis of ischemic stroke
Despite the association between elevated TGs and an increased risk of ischemic stroke, low serum TG levels do not necessarily benefit patients already suffering from acute stroke (Table 3). In a prospective study of 790 patients with AIS (men, 41.0%, age 79.4±6.8 years), the researchers found that the serum log-TG levels in patients with moderate/severe stroke at admission were significantly lower than those in patients with mild stroke (115±65 vs. 120±51, P<0.05) and serum log-TG levels of patients who died during hospitalization were significantly lower than those of discharged patients (P<0.001) [48]. In a binary logistic regression analysis, the serum log-TG levels were an independent predictor of moderate/severe stroke and nosocomial mortality at admission (RR, 0.24; 95% CI, 0.08 to 0.68; P<0.01 and RR, 0.09; 95% CI, 0.01 to 0.87; P<0.05) [48]. Dziedzic et al. [49] showed that patients with severe stroke on admission had significantly lower serum TG levels than patients with mild/moderate stroke on admission (1.04±0.60 mmol/L vs. 1.70±1.30 mmol/L). Moreover, after adjusting for age, sex, atrial fibrillation, diabetes mellitus, obesity, and ischemic heart disease, patients with TGs >2.30 mmol/L had a lower risk of severe stroke than those with TGs ≤2.30 mmol/L (OR, 0.58; 95% CI, 0.35 to 0.95) [49]. Similarly, an inverse relationship between National Institutes of Health Stroke Scale scores (NIHSS; with higher score meaning worse severity) and fasting TGs has been reported by several studies [50,51]. Higher fasting TGs (≥1.70 mmol/dL) within 24 hours after admission were also associated with a lower infarct volume on brain computed tomography (P=0.014) [52].

Effects of triglycerides on the severity and prognosis of ischemic stroke
Low TG levels may also have detrimental effects on long-term prognoses. In a study involving 1,464 patients with small artery occlusion, a subtype of ischemic stroke, the researchers found that elevated TG levels were a protective factor for death 36 months after stroke (RR, 0.05; 95% CI, 0.00 to 0.57) [53]. In a prospective study of 1,310 nondiabetic acute stroke patients with a mean follow-up time in survivors of 1,195 days, lower fasting TG levels independently predicted higher mortality, whereas serum cholesterol levels were not an independent predictor [51]. The relative hazard per additional quartile of TGs was 0.84 (95% CI, 0.77 to 0.91) [51]. Similarly, a recent study also showed that lower TG levels were related to higher all-death cause and vascular death rates in all patients and in the non-cardioembolic stroke group (P<0.001, both), but not in the cardioembolic stroke group [50]. In a Cox regression analysis with multivariate adjustment, compared with the highest quartile, the adjusted HR of the lowest quartile for all-cause death was 2.58 (95% CI, 1.38 to 4.82) and that for vascular death was 3.50 (95% CI, 1.39 to 8.82) in the non-cardioembolic stroke group [50].
Notably, the effects of TGs on the early prognosis are more complex. Most studies have found that hypertriglyceridemia is a protective factor against ischemic strokes. A hospital-based retrospective study with 530 patients with ischemic stroke lasting less than 7 days investigated the relationship between metabolic syndrome and the short-term prognosis of ischemic stroke. After adjusting for age and sex, a multiple logistic regression model analysis showed that elevated TGs decreased the risk of poor functional outcome at 30 or 90 days [54]. In a study evaluating the effect of TGs on the clinical outcome of patients with AIS after 3 months, the researchers included 1,006 AIS patients (median age 68.5 years; 58.2% men) admitted to the hospital between 2011 and 2014, used the modified Rankin Scale (mRS) score at 3 months after the attack to define the clinical endpoint (excellent outcome, mRS 0–1; good outcome, mRS 0–2; and death, mRS 6), and found that the TG levels were positively correlated with excellent outcome (P<0.001) and good outcome (P<0.001), but were negatively correlated with death (P<0.001) after adjusting for various confounding factors, based on the patient’s blood TG level at admission [55]. To evaluate the effect of the TG/HDL-C ratio on the mortality rate of AIS after 3 months, the researchers recruited 1,459 patients (median age, 68.5 years; men 58.5%) from 2011 to 2017 and divided them into retrospective training and prospective test cohorts. Multivariate analysis found that elevated TG levels were associated with a decreased risk of the 3-month mortality rate (HR, 0.42; 95% CI, 0.26 to 0.60; P<0.001 and HR, 0.47; 95% CI, 0.22 to 0.73; P<0.001, respectively) in both the training and test cohorts [56]. In a retrospective study, Kang et al. [57] enrolled 556 patients with ischemic stroke and tested their fasting and nonfasting TG levels on admission. A multivariate analysis showed that after adjusting for several confounding factors, including the use of statins after discharge, higher fasting TG levels and higher nonfasting TG levels were both significantly associated with good 3-month functional activity after AIS (OR, 2.93; 95% CI, 1.67 to 5.14 and OR, 2.66; 95% CI, 1.51 to 4.67, respectively) [57]. In a meta-analysis, researchers included 21 full-text studies from the Cochrane Library, PubMed, and Web of Science databases. The results showed that the serum TG levels were significantly correlated with the death of AIS patients (OR, 0.65; 95% CI, 0.43 to 0.98) [58]. A subgroup analysis showed that both univariate analyses and multivariate analyses indicated that higher TG levels could reduce the mortality of AIS patients (OR, 0.40; 95% CI, 0.28 to 0.57 and OR, 0.65; 95% CI, 0.43 to 0.98) [58].
The effects of TGs on an early prognosis remain unclear. In a cross-sectional observational study, researchers recruited 250 participants with AIS and found that TGs were a protective factor for very severe stroke (NIHSS ≥21) (OR, 0.55; 95% CI, 0.34 to 0.91; P=0.019) through univariate regression analysis. However, after adjusting for age and sex, no significant association was observed in multivariate logistic regression analysis (OR, 0.75; 95% CI, 0.43 to 1.30; P=0.306) [59]. In a meta-analysis, the summary of results from two studies indicated that TG levels were positively correlated with early neurological deterioration in patients with AIS (OR, 5.31; 95% CI, 1.79 to 15.70) [58]. Researchers also found that elevated TG levels were an independent risk factor for relapse 3 months after stroke (RR, 2.32; 95% CI, 1.10 to 4.89; P=0.027) [53]. In another study that aimed to uncover the relationship between dyslipidemia and the outcome (NIHSS >10 at discharge or death) of AIS, researchers included 1,568 patients with AIS from four hospitals from January 2006 to December 2008. Through a multivariate logistic regression analysis or propensity score-adjusted analysis, the results demonstrated that TG levels were not significantly associated with the outcome of AIS (OR, 0.88; 95% CI, 0.39 to 1.99 and OR, 1.12; 95% CI, 0.54 to 2.32, respectively) [60]. In another meta-analysis of cohort studies that investigated the correlation between metabolic syndrome and stroke recurrence, researchers found that patients with metabolic syndrome had a significantly increased risk of stroke recurrence (RR, 1.52; 95% CI, 1.17 to 1.97), although there was no obvious association between higher TG levels and stroke recurrence (RR, 1.04; 95% CI, 0.84 to 1.29) [61]. In addition, in genetically modified hypertriglyceridemia mice (ApoCIII transgene or GPI-HBP1 knockout), severe hypertriglyceridemia did not reduce ischemic brain damage [62]. These results suggested that the role of hypertriglyceridemia in poor early outcomes, such as relapse and death, of ischemic stroke is still depressing.
Effects of triglyceride reduction on ischemic stroke
Clinical trials assessing the efficacy of TG reduction in preventing ischemic strokes are limited and less convincing, mainly because of the lack of drugs targeting only TGs. The Veterans Affairs HDL Intervention Trial, a placebo-controlled, randomized trial conducted in 20 veteran affairs medical centers, studied the effects of gemfibrozil, a type of fibrate that increases HDL-C and reduces TGs in stroke prevention in men with coronary heart disease and low levels of both HDL-C and LDL-C (mean, 0.82 and 2.90 mmol/L, respectively) [63]. After a follow-up of up to 5 years, 134 participants developed stroke, 76 in the placebo group, and 58 in the gemfibrozil group, with 90% ischemic stroke. The adjusted RR reduction of gemfibrozil for stroke was 31% (95% CI, 0.02 to 0.52); additionally, the reduction in risk was evident after 6 to 12 months [63]. This was the first trial that showed lipid therapy aimed at raising HDL cholesterol and lowering TGs reduced the stroke incidence. The results, however, could not yield the recommendation of gemfibrozil for prevention of stroke because of the specificity of participants, nor could it prove the statement that reducing TGs could prevent stroke as gemfibrozil raised HDL-C and lowered TGs simultaneously. In another multicenter, randomized, double-blind, placebo-controlled clinical trial that involved 8,179 patients, researchers found that in those patients who had elevated fasting TG levels (1.69 to 5.63 mmol/L) and normal LDL-C levels (1.06 to 2.59 mmol/L) and had been taking statins steadily for at least 4 weeks, using 2 g of icosapent ethyl twice daily could reduce their TG levels (median decrease of 18.3% in the icosapent ethyl group vs. median increase of 2.2% in the placebo group) and significantly reduced the risk of ischemic events (HR, 0.75; 95% CI, 0.68 to 0.83; P<0.001) as well as nonfatal stroke (HR, 0.71; 95% CI, 0.54 to 0.94) [64].
To summarize, based on the current literature on the associations between TGs and stroke, it is highly likely that elevated TGs, especially nonfasting TGs, are a predictor of ischemic strokes. However, TGs may also exert protective effects during the occurrence, progression, and recovery of acute cerebral ischemia. The potential mechanisms behind these observations are reviewed in the next section.
Mechanisms behind hypertriglyceridemia and ischemic stroke
Atherosclerosis
Atherosclerosis is an underlying risk factor of ischemic stroke. Small plaques peeling off from unstable atherosclerotic plaques may embolize the intracranial arteries and cause artery-to-artery embolic stroke [65]. Atherosclerosis occurring in certain crucial vessels may also produce an in situ thrombus and cause ischemic stroke. Notably, asymptomatic carotid stenosis has been recognized as a well-documented risk factor for stroke, and prophylactic carotid endarterectomy can decrease the risk [3].
Despite lesser realization as an atherogenic factor than LDL-C, hypertriglyceridemia may promote the onset of ischemic stroke as it contributes to atherogenesis. There is a significant correlation between the carotid intima-media thickness and postprandial TG levels, implicating postprandial hypertriglyceridemia as an independent risk factor for early atherogenesis [66,67]. Theoretically, hypertriglyceridemia may lead to atherosclerosis via the following four mechanisms: subendothelial retention of remnant particles, toxic effects of lipolytic products, elicitation of endothelial dysfunction, and establishment of inflammation.
Subendothelial retention of remnant particles
Lipoproteins must enter the intima of arteries and be retained in the subendothelial space to cause atherosclerosis [68]. Nascent CM and even VLDL seem to be excluded from the intima due to their large size [69]. These factors might explain why the extremely high levels of TGs (>24.86 mmol/L), as seen in familial LPL deficiency with chylomicronemia syndrome, do not accelerate atherosclerosis [44]. However, Daugherty et al. [70] demonstrated the aortic accumulation of β-VLDL caused by diet-induced hypercholesterolemia in rabbits. Rapp et al. [71] demonstrated that human atherosclerotic plaques contained intact TRLs. In addition, Proctor and Mamo [72] demonstrated that small particles, such as CM remnants, could penetrate the arterial wall and were retained in the subendothelial space. Therefore, the remnant particles of CM and VLDL could penetrate into the intima of the arteries, become trapped, initiate inflammation, and trigger atherosclerosis, because they are smaller in size than CM and VLDL (Figure 2A) [68,73]. Furthermore, unlike LDL, remnant particles efflux very slowly from the intima, compared to their entry rate, and could be taken up directly by macrophages without modification [74], favoring the formation of foam cells and atherosclerotic lesions. Therefore, this mechanism is the most simple explanation for the association between elevated nonfasting TG levels and an increased risk of ischemic stroke, as nonfasting TG levels are a parameter reflecting the level of TG-rich remnants [44,45]. However, as TGs, but not cholesterol, could be degraded by macrophages in the intima, it might not be TGs but cholesterol inside the remnants that cause atherosclerosis [44]. There has been evidence demonstrating that high RLP-C levels are a significant and independent predictor of ischemic stroke events in patients with metabolic syndrome and mild carotid plaques [75]. Moreover, although TG-rich particles in the intima still contain more TGs than cholesterol, the amount of cholesterol in each of these particles is 5 to 20 times greater than that in LDL [76]. Therefore, the remnants have potential toxicities and proinflammatory activities on the vascular wall and can activate macrophages to secrete various cytokines, which may exacerbate the process of inflammation [74] and eventually lead to atherogenesis and ischemic stroke.
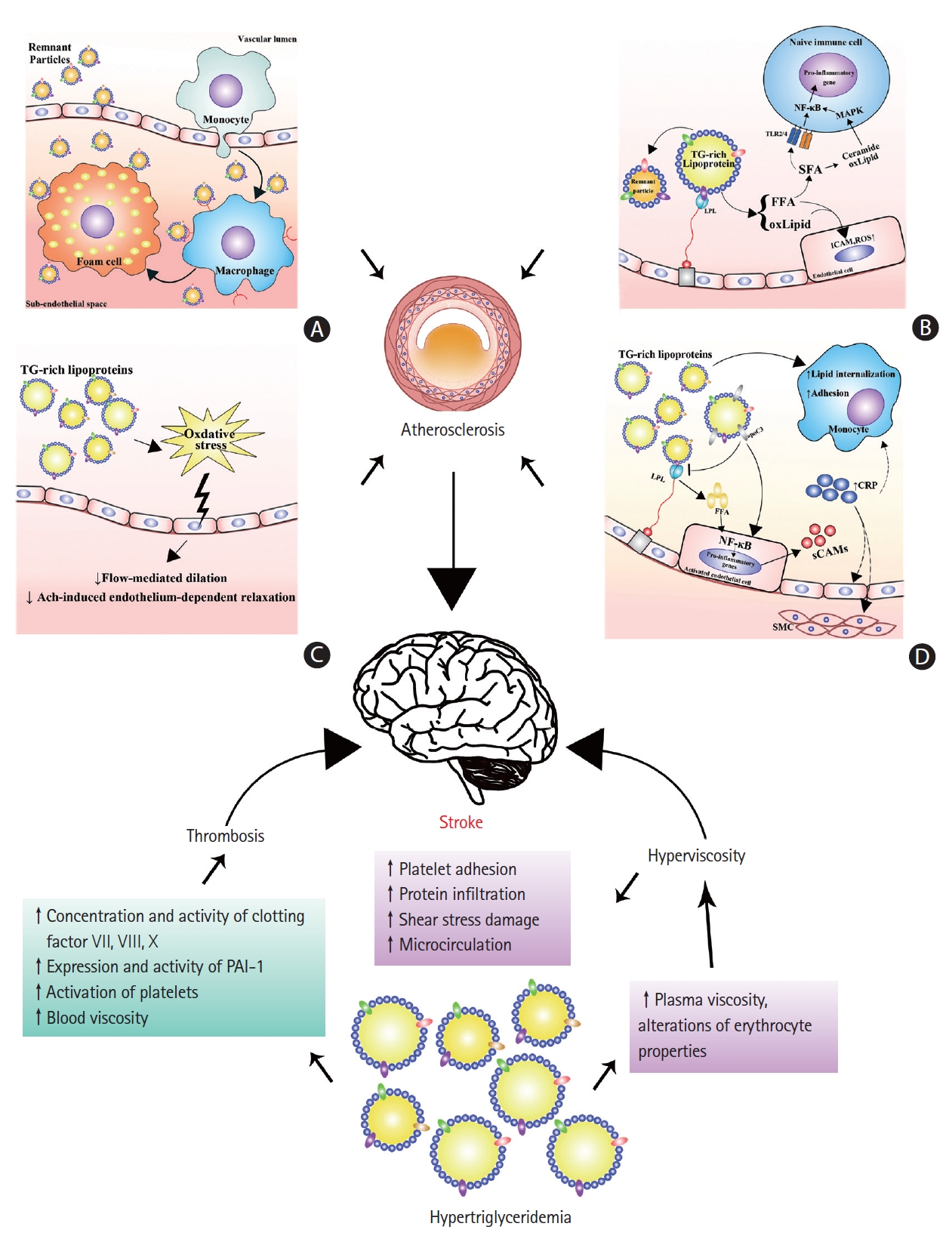
Mechanisms behind hypertriglyceridemia and ischemic stroke. Hypertriglyceridemia may increase the risk of ischemic stroke by predisposing patients to atherosclerosis, thrombosis, and hyperviscosity. Hypertriglyceridemia predisposes atherosclerosis mainly through the subendothelial deposition of remnant particles (A), toxic effects of triglyceride-rich lipoprotein (TRL) lipolytic products (B), impairment of endothelial function (C), and the establishment of local and systemic inflammation (D). Hypertriglyceridemia leads to increased concentrations and activity of clotting factors VII, VIII, X, and plasminogen activator inhibitor-1 (PAI-1), activation of platelets and higher blood viscosity, which together predict a higher risk of thrombosis. Hypertriglyceridemia represents a higher blood viscosity because of elevated plasma viscosity and altered erythrocyte properties. Hyperviscosity can increase platelet adhesion, protein infiltration to the subendothelial space, and shear stress damage, and impair microcirculation, leading to a higher risk of atherosclerosis, thrombosis, and cerebral ischemia. TG, triglyceride; NF-κB, nuclear factor κB; MAPK, mitogen-activated protein kinase; TLR, Toll-like receptor; SFA, saturated fatty acid; FFA, free fatty acid; oxLipid, oxidized phospholipid; ICAM, intracellular adhesion molecule; ROS, reactive oxygen species; LPL, lipoprotein lipase; CRP, C-reactive protein; sCAM, soluble cell adhesion molecule; SMC, smooth muscle cell.
Toxic effects of lipolytic products
In hypertriglyceridemia, LPL-mediated hydrolysis of TRLs may elevate the concentrations of free fatty acids (FFAs) in the proximity of the endothelium and in the systemic circulation, enhancing endothelial injury, inflammation, and atherosclerosis (Figure 2B) [74,77]. Lipolytic products from TRLs increase the endothelial permeability and induce apoptosis in human aortic endothelial cells (HAECs) [78]. Lipolysis of TRLs releases FFAs and oxidized lipids, such as oxidized phospholipids and 9- and 13-hydroxyocatadecadienoic acids (HODEs). The FFA fractions elicit proinflammatory responses and increase the expression of intracellular adhesion molecule (ICAM) and the production of reactive oxygen species (ROS) in HAECs [79]. In particular, saturated fatty acids (SFAs) may activate Toll-like receptor 2 or 4 (TLR2 or TLR4) in naive immune cells, leading to the activation of nuclear factor κB (NF-κB) and the subsequent transcription of proinflammatory genes [74]. In addition, both oxidized phospholipids and ceramide produced from SFAs may activate the mitogen-activated protein kinase (MAPK) signaling pathway and enhance the transcriptional effectiveness of NF-κB [74]. HODEs, especially 13-HODEs, are potent inducers of ROS production in HAECs [79]. HODEs lead to the synthesis of proinflammatory genes through NF-κB and/or other mechanisms [74]. Furthermore, excessive intracellular ROS production may cause fatty acid peroxidation and result in the accumulation of peroxides and oxidative damage to mitochondria, which in turn exacerbate oxidative stress [80]. Mitochondrial dysfunction can also result in vascular dysfunction and vascular cells apoptosis and favor the rupture of plaques and onset of stroke. Together, increased lipolytic products of TRLs in hypertriglyceridemia can increase the oxidative stress, cause endothelial injury, and elicit an inflammatory state, which may accelerate atherosclerosis and trigger ischemic stroke.
Elicitation of endothelial dysfunction
Endothelial dysfunction is considered an early feature of atherosclerosis [81]. Hypertriglyceridemia, especially postprandial hypertriglyceridemia, has been shown to be associated with endothelial dysfunction in several studies (Figure 2C). Transient impairment of flow-mediated dilation (FMD) in the brachial artery has been observed in healthy subjects with transient hypertriglyceridemia induced by either intravenous or oral fat challenge [82,83]. However, transient postprandial hypertriglyceridemia does not impair the endothelial function in forearm resistance vessels [84]. Consistent results of impaired FMD and acetylcholine-induced endothelium-dependent relaxation have been reported in patients with hypertriglyceridemia [81,85,86]. Reduction in serum TG levels via pharmacological therapy improved the endothelial function in obese subjects (men) and patients with coronary heart disease [86,87]. Oxidative stress has been implicated as a result of postprandial hypertriglyceridemia and a cause of endothelial dysfunction. Increased leukocyte O2- production is associated with elevated serum TGs and impaired FMD after a high-fat meal [88]. Lipid-derived free radicals also have been observed to increase after a fat tolerance test, and the magnitude is greater in patients with type 2 diabetes than in healthy individuals [89]. Remnants are also associated with endothelial cell apoptosis. High concentrations of CM remnants induce endothelial cell apoptosis. RLP induces apoptosis in endothelial cells by NAD(P)H oxidase-mediated superoxide and by cytokine production via lectin-like oxidized low-density lipoprotein receptor-1 (LOX-1) [90]. Collectively, hypertriglyceridemia can induce endothelial dysfunction, probably by increasing oxidative stress and inducing apoptosis, thus promoting atherosclerosis and ischemic strokes.
Establishment of inflammation
Hypertriglyceridemia may lead to a systemic inflammatory state via the activation of endothelial cells and leukocytes and elevation of CRP, thereby accelerating atherosclerosis (Figure 2D). The critical steps of inflammation in atherosclerosis involve the adhesion of monocytes to endothelial cells and their subsequent transmigration into the vascular wall. The plasma levels of soluble cell adhesion molecules (sCAMs) can reflect the degree of endothelial activation [91]. Increased levels of soluble vascular cell adhesion molecule-1 (VCAM-1), ICAM-1, and E-selectin, as well as leukocyte cell adhesion molecules CD18, CD49d, and CD54 are observed in patients with hypertriglyceridemia [91,92]. Chronic low-dose lipid infusion in healthy individuals also leads to the induction of soluble ICAM-1, VCAM-1, and endothelin-1 (ET-1), which is independent of its insulin-desensitizing effects [93]. Monocyte chemoattractant protein-1 (MCP-1), a C-C chemokine, stimulates the migration of monocytes through arterial endothelial layers and is postulated to play a critical role in the development of atherosclerosis [94]. Maeno et al. [95] reported that IDL can stimulate MCP-1 mRNA expression in cultured human umbilical vein endothelial cells (HUVECs) [95]. In addition, RLPs can induce MCP-1 expression in HUVECs [96]. Others have demonstrated that rat CM remnants strongly stimulate mRNA expression and protein secretion of MCP-1 in vascular smooth muscle cells by activating p38 MAPK [97]. Prandial hypertriglyceridemia also transiently increases the lipid internalization of monocytes and their adhesion to VCAM-1 from healthy subjects fed a standardized high-fat meal [98]. In addition, when cultured in the presence of RLPs, the adhesion of human monocytic U937 cells to HUVECs increased under physiological laminar flow conditions [99]. These results favor the recruitment of monocytes during the development of atherosclerosis. Mechanistically, VLDL can activate NF-κB in endothelial cells, which is mediated by the release of VLDL fatty acids but not by the oxidation of VLDL [100]. FFAs can also activate NF-κB in endothelial cells and regulate the TNF-α-induced activation of endothelial cells [100,101].
Another potential mechanism linking hypertriglyceridemia to atherosclerosis is related to CRP [102]. Elevated CRP levels also correlate with elevated TG levels and an increased risk of coronary heart disease [102,103]; however, it remains unknown whether it is a causal factor in the pathogenesis [103,104]. Nevertheless, a series of experimental studies indicated that CRP promotes atherothrombosis via the regulation of endothelial cells, alterations in the functions of vascular smooth muscle and monocytes/macrophages, changes in matrix biology, and the promotion of coagulation [105]. In a rat model of middle cerebral artery occlusion, human CRP infusion could increase the infarct volume, indicating that human CRP contributes to ischemic tissue damage [106]. Overall, the proinflammatory state elicited by hypertriglyceridemia may contribute to the pathogenesis of atherosclerosis and even alter the pathophysiology of ischemic stroke.
Hyperviscosity and thrombosis
Blood viscosity is closely associated with plasma viscosity and erythrocyte properties (i.e., erythrocyte concentration, volume, deformability, and aggregation), with the latter accounting for a larger part [107,108]. An association between increased plasma TGs and elevated blood viscosity was reported previously [109,110]. The most straightforward explanation for this phenomenon may be that the elevated plasma viscosity is caused by increased levels of lipoproteins, as the plasma viscosity is determined by the water content and macromolecular components [107,108]. Indeed, in vitro studies have shown that isolated CM, VLDL, and LDL cause a dose-dependent and exponential rise in plasma viscosity, which is greater with larger lipoproteins [111,112]. Moreover, in patients with metabolic syndromes, hypertriglyceridemia also independently predicts erythrocyte hyperaggregability [110]. Consistently, an LPL-deficient mouse model with severe hypertriglyceridemia also displays an erythrocyte disturbance, which includes decreased deformability, electrophoresis rate, membrane fluidity, and increased osmotic fragility [109]. Therefore, hypertriglyceridemia may additionally cause high blood viscosity via alterations of erythrocyte properties (Figure 2).
Blood viscosity and its determinants (i.e., plasma viscosity, fibrinogen, and hematocrit) are associated with carotid intima-media thickness, an early stage of atherosclerosis in the Edinburgh Artery Study [113]. Additionally, in the same study, elevated blood viscosity, hematocrit, plasma viscosity, and fibrinogen were shown to correlate with stroke occurrence [114]. In addition, worsened blood rheological properties were observed in patients with stroke [115], who showed enhanced whole blood viscosity due to the substantial increase in plasma viscosity and the impairment of microrheological blood properties—that is, elevated erythrocyte aggregability and decreased deformability compared with the healthy group [115].
Elevated blood viscosity may promote atherosclerotic development by increasing the platelet adhesion to the sub-endothelium and protein infiltration into the arterial wall, and altering local shear forces at the sites of atherogenesis [113]. Elevated plasma viscosity may also lead to atherothrombosis due to impaired microcirculatory flow, increased shear stress damage at the blood-endothelial interface, enhanced interactions of plasma protein with the endothelium in post-stenotic recirculation zones, and an increased propensity of thrombosis [116]. In summary, hypertriglyceridemia may lead to the elevation of blood viscosity by increasing the plasma viscosity and causing erythrocyte disturbance, which can promote atherosclerosis, thrombosis, and cerebral ischemia, thus increasing the risk of ischemic stroke.
Thrombosis is a critical event in arterial diseases associated with ischemic stroke [117], as occlusion of intracranial arteries by either distant emboli (e.g., cardioemboli or carotid atherosclerotic plaques) or in situ thrombi can result in the onset of ischemic stroke [65]. Hypertriglyceridemia may represent a procoagulant state and therefore contribute to the onset of ischemic stroke through its effects on thrombosis (Figure 2) [102].
Hypercoagulability and reduced fibrinolytic capacity are associated with hypertriglyceridemia and postprandial triglyceridemia [118-120]. Elevations in the concentrations and activity of clotting factors VII, VIII, and X have been implicated in hypertriglyceridemia [118,121-123]. In particular, Factor VII, an essential component in the extrinsic coagulant pathway, is closely associated with hypertriglyceridemia [121-124]. Notably, the activity of FVII is an independent predictor of myocardial infarction in healthy middle-aged men with fatal coronary events [120]. However, activation of the intrinsic pathway of coagulation does not contribute to the hemostatic imbalance in hypertriglyceridemia [125]. In addition, the increased expression and activity of plasminogen activator inhibitor-1 (PAI-1) is correlated with an increase in plasma TGs [119,122,126,127]. As PAI-1 is the major physiological inhibitor of plasminogen activators in the circulation and the principal inhibitor of the fibrinolytic system [120], the elevation of PAI-1 indicates a suppression of fibrinolytic capacity and thus a high risk of thrombosis. Notably, the dietary intake of unsaturated fatty acids, rather than SFAs, is more potent in elevating the concentrations and activity of FVII and PAI-1 [128,129]. In addition, elevated tissue-type plasminogen activator and antithrombin-III have also been observed in patients with hypertriglyceridemia [123,126,127], which may be a protective mechanism against thrombotic events in these patients.
Regarding the molecular mechanisms of hypercoagulability and hypertriglyceridemia, FVII activation by TRL is dependent on LPL-mediated lipolysis [130]. VLDL from normal or hyperlipidemic individuals can stimulate the secretion of PAI-1 from HUVECs [131,132]. VLDL also enhances the synthesis and release of PAI-1 in HepG2 cells [133], which occurs through the binding of VLDL particles to LDLRs on the cells [131,133]. CM remnants increased the production of PAI-1 in endothelial cells via the MAPK pathway and redox system [134]. Notably, inhibition of the renin-angiotensin system with angiotensin-converting enzyme inhibitors and angiotensin type 1 receptor blockers (ARBs) could reduce the production of PAI-1 by remnants in endothelial cells [134]. In addition, patients with primary hypertriglyceridemia have an increased activation of platelets in vivo [135], which may be related to changes in the lipid composition of the platelet membranes, oxidative stress, and the potential activation effects of VLDL on platelets [135].
In conclusion, hypertriglyceridemia may promote thrombosis and the onset of ischemic stroke through a procoagulant effect involved in the disturbance of both blood coagulation and fibrinolysis [120,136]. Hyperviscosity may also contribute to hypercoagulability, which is discussed in the next section.
Protective role of triglycerides in acute ischemic stroke
Although hypertriglyceridemia may increase the risk of ischemic stroke by promoting atherosclerosis, thrombosis, and hyperviscosity, low serum TGs also appear to be detrimental to patients with respect to the severity on admission and outcome after acute stroke [49-52]. These notable clinical findings raise the question of whether TGs are protective against cerebral ischemic injury. Although it remains unclear, several mechanisms may be considered. First, low serum TG levels may reflect the underlying poor nutritional status [49-51]. Although malnutrition after stroke is a risk factor for poor outcomes, it could not explain the severity of stroke on admission [49]. Additionally, the association between TG concentrations in plasma and stroke mortality is independent of albumin and hemoglobin, which are two parameters reflecting the nutritional status [50]. These results imply that TGs might confer additional effects during and after stroke occurrence. Another plausible mechanism for the inverse correlation between TGs and stroke severity and outcomes focuses on the protective function of TGs [50]. Cellular accumulation of excessive fatty acids in TG pools likely diverts these molecules from pathways that lead to cytotoxicity. Therefore, intracellular TGs may serve as a buffer against lipotoxicity [137]. In this case, unsaturated fatty acids can exert a protective effect against lipotoxicity through the promotion of TG accumulation. Mitochondrial adaptations may be a third explanation. Livers from hypertriglyceridemic mice have a greater content of glycerolipids and an increased oxidation of FFAs in the mitochondria. However, mitochondria in hypertriglyceridemic livers have a high resting respiration rate but normal oxidative efficiency of phosphorylation, reducing the generation of ROS and maintaining a normal metabolic phenotype; therefore, mitochondria do not develop insulin resistance [138]. The brain contains high concentrations of polyunsaturated fatty acids that are susceptible to lipid peroxidation, consume relatively large amounts of oxygen for energy production, and have lower antioxidant defenses than other organs [139]. As a result, the brain is believed to be particularly vulnerable to oxidative stress. However, we hypothesize that under chronic hypertriglyceridemia, the brain might develop compensatory mechanisms (e.g., mitochondrial adaptations) against an abnormal metabolic status that likely exerts protective functions during or after ischemic stroke.
Conclusions and future prospects
As an increasing number of well-conducted studies have unveiled the relationship between plasma TGs and the risk of ischemic stroke [43,44,140-142], we believe that hypertriglyceridemia is a neglected risk factor for ischemic stroke. In this review, we have summarized the possible mechanisms by which hypertriglyceridemia is related to ischemic stroke, including the increased propensity for atherogenesis and thrombosis and the status of blood hyperviscosity. Notably, these three mechanisms are not exclusive and crosstalk with each other. However, there is a huge gap in the interpretation of rare and scattered experimental studies with observations from large prospective clinical studies. Therefore, further targeted and systemic studies are needed to confirm the role of hypertriglyceridemia in promoting the onset of ischemic stroke. Clinically, because nonfasting TGs are more strongly associated with ischemic stroke (as well as other cardiovascular diseases) than fasting TGs [44,45,143], the standardized measurement of nonfasting TGs to predict the risk of cardiovascular diseases may be more reasonable [44,143,144]. In addition, the role of hypertriglyceridemia during and after the period of stroke is unclear and requires further research. Given the inverse correlation between TG levels and stroke outcomes [49-52], it is important to determine whether TGs play a protective role in patients with stroke, which may be important for clinical care and secondary prevention.
Notes
Disclosure
The authors have no financial conflicts of interest.
Acknowledgements
This work was supported by the National Key Research and Development Program (2019YFE0113500) and the National Natural Science Foundation of China (No. 81770268 and U1803125).