Introduction
Malignant cerebral edema following ischemic stroke is life threatening. The pathophysiology of brain edema involves failure of the sodium-potassium adenosine triphosphatase pump and disruption of the blood-brain barrier, leading to cytotoxic edema and cellular death.1 The Monro-Kellie doctrine dictates that since the brain is encased in a finite space, increased intracranial pressure (ICP) due to cerebral edema can result in herniation through the foramen magnum and openings formed by the falx and tentorium.2 Moreover, elevated ICP can cause secondary brain ischemia through decreased cerebral perfusion and blood flow, brain tissue hypoxia, and metabolic crisis.3 Direct cerebrovascular compression caused by brain tissue shifting can lead to secondary infarction, especially in the territories of the anterior and posterior cerebral artery.4 Tissue shifts can also stretch and tear cerebral vessels, causing intracranial hemorrhage such as Duret's hemorrhage of the brainstem.4 Physicians treating ischemic stroke with swelling should utilize every available medical and surgical therapy to minimize secondary brain injuries. Therefore, patients with space-occupying ischemic stroke are advised to be admitted to an intensive care unit (ICU) for constant neurological monitoring.5
Physicians who manage stroke patients should try to understand the body as a whole and not be limited to the brain. It is possible that provision of comprehensive care is the critical factor that determines patient outcomes, rather than focusing on one specific therapy. However, managing the brain and the rest of the body simultaneously presents a challenge to the physician, as focusing on one organ may be at the cost of another.
In this review, we provide a comprehensive overview of the management of cerebral and cerebellar infarction with malignant edema. We discuss the measurement of ICP and strategies to lower ICP, including decompressive craniectomy and therapeutic hypothermia. Finally, we discuss ventilatory support and the advantages and limitations of a sedation-off wake-up test.
Measurement of ICP
ICP is the pressure exerted by the brain, blood, and cerebrospinal fluid (CSF) in the intracranial vault. The skull is a rigid container filled with the brain, blood, and CSF. Expansion of one component occurs at the expense of others, with increases in ICP leading to intracranial hypertension. Normal ICP values for adults are 7-15 mmHg.6 Generally, ICP values greater than 20 mmHg require treatment.7 However, ICP should be interpreted in the context of cerebral perfusion pressure (CPP). CPP is a major factor that affects cerebral blood flow (CBF). It is determined by calculating the difference between mean arterial pressure (MAP) and ICP (CPP = MAP-ICP). CPP can therefore decrease as a result of decreased MAP, increased ICP, or a combination of both. CPP is proportional to CBF as long as cerebrovascular resistance remains constant. To avoid secondary brain ischemia after stroke, extremely low CPP should be avoided. Optimal CPP values have not been clearly established, but 50-60 mmHg is generally accepted as the minimum pressure required to prevent further brain injury.3
The gold standard assessment of ICP requires insertion of an extraventricular drain (EVD) into the lateral ventricle with a connection to an external pressure transducer.8 In addition to monitoring absolute pressure values and waveforms, an EVD catheter allows CSF diversion to reduce ICP. Alternatively, ICP can be measured through intraparenchymal, transducer-tipped catheters, including fiber optic and microstrain-gauge sensors.8 A parenchymal catheter can provide continuous measurement of ICP and allows analysis of the interaction between ICP and MAP (pressure reactivity index) as well as real-time values and ICP waveforms.9 Although this device provides a quantitative value for ICP, it can only be calibrated prior to placement of an ICP probe and is vulnerable to drift.
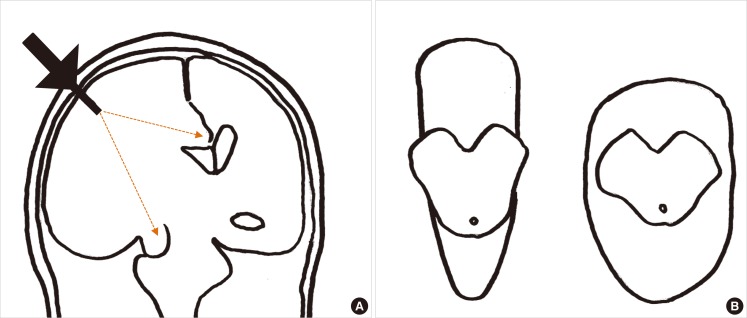
There are controversies surrounding ICP monitoring in patients with stroke. Early studies showed that ICP monitoring was useful for predicting clinical outcomes after acute hemispheric stroke. ICP often correlated with clinical deterioration, final outcome, and computed tomography findings.10 However, a subsequent study of patients with malignant middle cerebral artery (MCA) infarctions showed that pupillary abnormalities and signs of severe brainstem compression were sometimes present despite normal ICP.11 Randomized clinical trials of ICP monitoring have not been performed in patients with stroke. A randomized clinical trial of ICP monitoring in patients with traumatic brain injury failed to show superior efficacy of ICP monitoring over serial neurological examinations and repeated neuroimaging studies.12 Limitations of ICP monitoring in predicting neurological deterioration may be due to the distance between the site of ICP probe insertion and the site of herniation (Figure 1A), because the pressure gradient force is inversely proportional to the distance. The relative positioning of the tentorial aperture and the brainstem may also explain variations in herniation syndromes in patients with similar ICP values and intracranial pathological conditions. Some patients exhibit a narrow tentorial opening and a high degree of contiguity between the brainstem and the tentorial edge, whereas others exhibit a wide tentorial opening and perimesencephalic space. Such morphometric variations in the tentorial aperture and its regional anatomy may play a role in the varying clinical manifestations (Figure 1B).13 Alternatively, since the brain has compartments divided by hard dural structures, i.e., the falx cerebri and tentorium cerebelli, ICP probes placed in the right frontal cortex may not appropriately reflect the pressure in the left hemisphere or infratentorium (compartmentalized ICP).10,14 Malfunction of a mechanical device and probe drift may also contribute to inaccurate ICP values. Therefore, routine ICP monitoring without careful interpretation, neurological examination, and a neuroimaging study cannot be recommended in patients with cerebral and cerebellar infarct with swelling.5 However, it may be helpful in comatose patients especially during barbiturate coma therapy and ventilator care under heavy sedation because in these situations neurological examinations are not sensitive enough to detect secondary brain injury, and constant neuroimaging studies are not available. Multimodal neuromonitoring that includes ICP, CPP, CBF, brain oxygen tension, markers of brain metabolism (glucose, lactate, and pyruvate) and electroencephalography may be useful to further understand the physiology of comatose stroke patients.15
Medical management of intracranial hypertension
In general, there are four causes of intracranial hypertension following ischemic stroke: (1) brain swelling due to cytotoxic or vasogenic edema; (2) increased cerebral blood volume due to dilated cerebral arteries; (3) obstruction of venous outflow; and (4) acute hydrocephalus.16 The optimal approach to manage intracranial hypertension differs according to the underlying cause. Brain swelling requires osmotherapy and/or decompressive craniectomy, cerebral arterial dilatation requires a vasoconstriction stimulus such as hyperventilation, obstruction of venous outflow requires a revision of head position and loosening of tube ties (in cases not complicated by venous sinus thrombosis), and acute hydrocephalus can be managed with CSF drainage through an EVD.16
Our stepwise protocol for controlling ICP is outlined in Table 1. It starts with consideration of decompressive craniectomy and CSF drainage through an EVD because these are the most effective measures for ICP control.17 Details on these surgical options are described in subsequent sections.
General management
Assessment and management of the airway, breathing, and circulation is the initial step for treating increased ICP in unconscious stroke patients. Early rapid sequence intubation (RSI) should be considered for patients with comatose mental status and intracranial hypertension, and endotracheal intubation should be performed with medications to blunt any increase in ICP. Details on intubation, ventilatory support, and sedation are described in subsequent sections. Hypoxia and hypotension should be avoided during or after the procedure. Head elevation to 30¬į has been shown to reduce ICP, although this can be accompanied by a decrease in CPP that offsets the beneficial effects.18 The head should be maintained in a midline position and tight ties around the neck should be avoided to improve jugular venous drainage. Other general measures to treat intracranial hypertension include avoiding hypoxia, hypercapnia and hypotonic conditions, controlling fever and hyperglycemia, and treating seizures. Details on ventilatory support and the strategy for sedation and analgesia are described in subsequent sections.
Hyperventilation
Decreasing PaCO2 to 30-35 mmHg is an effective and rapid means to reduce ICP. A decrease in PaCO2 causes vasoconstriction, which lowers cerebral blood volume and thus lowers ICP. The effect is almost immediate but generally lasts only a few hours because the pH of CSF rapidly equilibrates to the new PaCO2 level.3 However, the effect of hyperventilation may be sustained for days in patients with excessive vasodilation and cerebral hyperemia.19 Prolonged, aggressive hyperventilation can cause cerebral ischemia in patients with brain injury,20 and the routine application of extreme hyperventilation is therefore generally considered harmful. Thus, the use of hyperventilation is best reserved for temporary, rescue therapy for sudden increases in ICP. It is not recommended to routinely hyperventilate patients for many hours or days.
CPP optimization
The minimal CPP required to prevent brain ischemia is generally accepted as 50-60 mmHg.3 However, there are two different approaches to whether CPP should be maintained at a higher or lower level. The Rosner concept advocates increasing MAP and targeting a higher CPP to maintain adequate CBF,21 whereas the Lund concept advocates decreasing resistance and intravascular hydrostatic pressure, and reducing cerebral blood volume, thereby increasing CBF and accepting a lower CPP.22 Recently, new approaches to individualize optimal CPP using multimodal neuromonitoring measures and statistics from these measures, such as the pressure reactivity index that is calculated using the moving correlation coefficient between ICP and MAP and the oxygen reactivity index that is calculated using the moving correlation coefficient between CPP and brain tissue oxygen, have been introduced.9,23 Details on different conceptual approaches are beyond the scope of this review.
Osmotherapy
Mannitol has been the cornerstone of osmotherapy for the treatment of intracranial hypertension. However, optimal usage of mannitol for stroke patients is uncertain. Usually, this drug is administered intravenously at a dose of 0.25-2.0 g/kg per requirement or every 6 hours. Serum osmolality up to 360 mOsm/kg can be tolerated.3 During osmotherapy with mannitol, it is crucial to maintain a hyperosmotic euvolemic state through adequate fluid balance. If not, the patient could suffer various adverse effects including dehydration, hypovolemia, hypotension, increased ICP, and decreased CPP, in addition to acute kidney injury and electrolyte imbalance. When mannitol needs to be discontinued after repeated use for several days, potential rebound increases in ICP should be monitored with consideration of mannitol tapering. Therapeutic drug monitoring for mannitol is unavailable in most hospitals, but physicians can use the osmolar gap, i.e., the difference between measured serum osmolality and calculated serum osmolarity, as an indirect indicator of serum mannitol levels. There are several approaches to calculate serum osmolarity, but the most simple, frequently used formula is as follows: serum osmolarity=(2√óNa)+(glucose/18)+(blood urea nitrogen/2.8). The osmolar gap correlates better than serum osmolality with the serum concentration of mannitol.24 When the osmolar gap is higher than 20 mOsm/L, mannitol is generally withheld to avoid accumulation in the brain and circulation.3 However, 55 mOsm/L has also been suggested as a threshold value.25
Hypertonic saline is an alternative to mannitol for osmotherapy for intracranial hypertension. Although hypertonic saline provides an osmotic effect similar to mannitol, it has a better reflection coefficient (1.0) than mannitol (0.9), and is therefore less able to cross the blood-brain barrier and may have a stronger osmotic effect. Additionally, hypertonic saline normalizes resting membrane potential and has an anti-inflammatory effect. In existing reports, the concentration of hypertonic saline used to treat intracranial hypertension was 1.7%-30%. A recent meta-analysis of five clinical trials showed greater ICP control with hypertonic saline than with mannitol in patients with traumatic brain injury (odds ratio, 1.16; 95% confidence interval, 1.00-1.33).17 Another recent meta-analysis of six studies showed that the percent decrease in ICP from baseline to either 60 minutes or the nadir after administration of 23.4% saline was 55.6% (standard error, 5.90; 95% confidence interval, 43.99-67.12).26 Possible adverse effects of hypertonic saline are rebound cerebral edema, hyperchloremic metabolic acidosis, phlebitis, congestive heart failure, transient hypotension, hemolysis, hypokalemia, renal failure, osmotic demyelination, subdural hemorrhage, seizures, and muscle twitching.27 Various methods have been used to administer hypertonic saline. At the Asan Medical Center, a 60-120 mL dose of 11.7% saline, the highest available osmolar agent in Korea, is administered over 10-20 minutes via a peripherally inserted central catheter as required or every 6 hours. Serum sodium levels up to 155-160 mEq/L are typically well tolerated.
Barbiturate coma therapy
Barbiturate coma therapy is reserved for patients with refractory intracranial hypertension and can be delivered using pentobarbital or thiopental.28 These drugs are titrated on the basis of ICP measurements and continuous electroencephalogram monitoring. Pentobarbital is administered at a loading dose of 5 mg/kg over 15-30 minutes (up to 50 mg/min) and then continuously infused at 1-5 mg/kg/h. Barbiturate coma therapy requires electroencephalograms to be continuously recorded and the barbiturate titration usually targets a burst-suppression pattern with approximately 6-8 seconds interbursts.19 Side effects of pentobarbital and thiopental include hypotension, myocardial suppression, respiratory suppression, infections, hepatic and renal dysfunction, thrombocytopenia, metabolic acidosis and gastric stasis. However, randomized clinical trials on the effect of barbiturate therapy for uncontrolled ICP have not been performed in stroke patients. Barbiturate coma therapy is the last resort for treatment of refractory intracranial hypertension due to a lack of sufficient evidence and various systemic side effects associated with it, including cardiac depression, arterial hypotension, and increased risk of infection.29
Surgical management of brain swelling
Medical management of brain edema and increased ICP may not be successful when significant swelling occurs in the cerebral or cerebellar hemisphere. Therefore, a decompressive craniectomy should be considered to relieve the mass effect of the swollen cerebral or cerebellar hemisphere on the thalamus, brainstem, and network projections to the cortex, manifested predominantly by decreased levels of arousal.5
Malignant MCA infarction
Malignant MCA infarction was originally defined as an acute infarction in the entire MCA territory evident on a computed tomography scan within the first 48 hours after symptom onset, with or without involvement of other vascular (anterior or posterior cerebral artery) territories.30 Subsequently, the term has been used to refer to large hemispheric infarcts that have occurred as a result of occlusion of the proximal MCA or internal carotid artery, with variable definitions (e.g., National Institutes of Health Stroke Scale score ‚Č•15-20; brain computed tomography ischemic signs involving >50%-66% of the MCA territory; and diffusion-weighted imaging infarct volume >145 cm3).31,32,33,34,35 Malignant MCA infarction accounts for approximately 5% of ischemic strokes and is most commonly caused by cardioembolism.30,36 Despite the best medical treatment, most patients deteriorate between post-ictus days 2 and 5, and malignant MCA infarction is characterized by severe morbidity and high mortality.30,37 Mortality ranges up to 70%-80% with medical treatment.38 For patients who present outside thrombolysis time windows or who already have areas of marked low density on computed tomography scans, treatment should aim to minimize brain swelling and control ICP. Among the variety of treatment modalities available to treat an increase in ICP, the most effective method is decompressive hemicraniectomy.17 In a meta-analysis of studies into the control of increased ICP, decompressive hemicraniectomy was found to cause a 19 mmHg mean reduction in pressure, which was superior to the reduction in ICP seen with the use of hyperventilation (6 mmHg), mannitol (8 mmHg), barbiturates (8 mmHg), hypothermia (10 mmHg), hypertonic saline (15 mmHg), or CSF drainage (15 mmHg).17
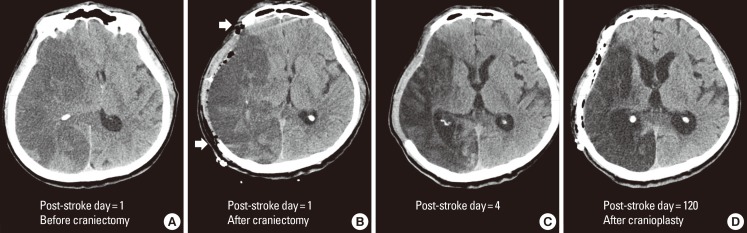
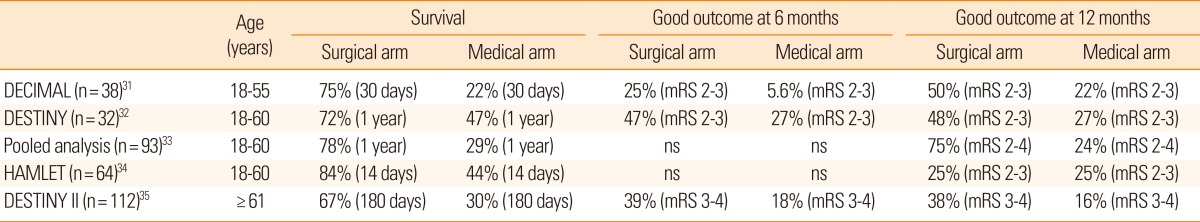
In decompressive hemicraniectomy, a bone flap over the frontal, temporal, parietal, and occipital lobe at the site of the infarct is removed, allowing the swollen brain tissue to expand extracranially (Figure 2). Decompressive hemicraniectomy with duroplasty can relieve horizontal and vertical tissue shifts, reduce ICP, improve CPP, and CBF, and alleviate vascular compression. During the last decade, several randomized clinical trials have shown that this procedure is most effective when performed within 48 hours post-ictus (Table 2).31,32,33,34 A hemicraniectomy performed too soon after ictus may result in unnecessary surgical procedures and complications, whereas a hemicraniectomy performed after herniation signs are fully developed may be of little value. Multiple randomized clinical trials have investigated if early (<48 hours post-ictus) hemicraniectomy improves clinical outcomes for patients with malignant MCA infarction. Trials included young patients (age ‚ȧ60 years) with severe symptoms (National Institutes of Health Stroke Scale score ‚Č•20 for the dominant hemisphere and ‚Č•15 for the nondominant hemisphere) and a decreased level of consciousness (score ‚Č•1 on item 1a of the National Institutes of Health Stroke Scale score or a gradual decrease in consciousness to a score ‚ȧ13 on the Glasgow coma scale for right-sided lesions or ‚ȧ9 for left-sided lesions). In a pooled analysis of three European trials, the number needed to treat for survival at 1 year was two and the number needed to treat for independent walking (modified Rankin Scale (mRS) score ‚ȧ3) was four.33
Despite unequivocal effects on survival, there has been criticism that hemicraniectomy can barely avert death to a vegetative or minimally conscious state. However, patients with malignant MCA infarctions who may have been in a vegetative state (mRS score=5) without hemicraniectomy could walk with some help (mRS=4) after hemicraniectomy. A recent meta-analysis showed that the vast majority of patients who underwent hemicraniectomy and their caregivers were satisfied with the outcome of surgery and would provide consent again for the procedure despite the high rate of physical disability and depression.39 Patients with an infarct in the dominant hemisphere may have a lower quality of life than patients with an infarct in the nondominant hemisphere because they have more severe language impairment. However, a meta-analysis found no differences in functional outcome between right- and left-hemispheric infarcts.40
Until recently, the efficacy of decompressive hemicraniectomy in patients 60 years of age or older has been uncertain. A recent randomized clinical trial showed that malignant MCA infarction patients aged >60 years who were treated with early (<48 hours post-ictus) hemicraniectomy had a higher survival rate and better functional outcome than patients who were managed conservatively (Table 2).35 Thus, old age per se should not be regarded as an exclusion criterion for hemicraniectomy after malignant MCA infarction. Preemptive decompressive hemicraniectomy is an effective treatment for brain swelling in patients with malignant MCA infarction.
Cerebellar infarction
A space-occupying cerebellar infarct may cause brain death through brainstem compression and obstructive hydrocephalus. Large infarct size on initial magnetic resonance images, mostly in the territories of the posterior inferior cerebellar artery and superior cerebellar artery rather than the anterior inferior cerebellar artery, can suggest poor prognosis.41 Consciousness typically deteriorates between post-ictus days 2 and 4,42 and surgical options should be considered when maximal medical therapy is failing.
One surgical option for treating a space-occupying cerebellar infarct is a suboccipital craniectomy, where the skull is removed and the dura expanded to relieve ICP caused by the swollen brain tissue (Figure 3). Although the efficacy of suboccipital craniectomy has been reported in observational and retrospective studies,42,43,44 no randomized clinical trials have been performed to test the efficacy of this procedure. The value of preemptive surgery (early surgery based on radiological findings such as cerebellar edema and hydrocephalus in a clinically stable patient) and the best neurosurgical approach (removal of necrotic tissue vs. decompression alone vs. decompression plus ventriculostomy) are unknown.5,45,46 In patients with acute hydrocephalus following cerebellar infarction, placement of an EVD (ventriculostomy) should be accompanied by suboccipital craniectomy.5
Therapeutic hypothermia
Clinical studies on therapeutic hypothermia have investigated the effects on two different outcomes: neuroprotection and ICP control. Therapeutic hypothermia is potentially neuroprotective through multiple mechanisms. Hypothermia reduces cerebral metabolism and consumption of oxygen and glucose by the brain, thereby preventing the failure of sodium transport and calcium influx, and decreasing the risk of cell death.47 Additional beneficial effects of hypothermia include the inhibition of excitatory neurotransmitters, free radicals, inflammation, and apoptosis. Reduced ICP is a robust manifestation of hypothermia and is driven by reductions in cerebral blood volume, vasogenic edema and inflammation, and mitigation of blood-brain barrier leakage.47
In stroke patients, therapeutic effects of hypothermia are equivocal despite robust benefits in animal models and preclinical trials. Small pilot studies suggest possible benefits of therapeutic hypothermia in stroke (Table 3) and observational studies suggest that therapeutic hypothermia is a potent anti-edema strategy. Induced hypothermia with a target temperature of 32-33‚ĄÉ helped control ICP elevation due to cerebral edema in patients with malignant MCA infarction,48,49 and induced hypothermia with a target temperature of 34.5‚ĄÉ reduced cerebral edema and hemorrhagic transformation after successful recanalization for acute ischemic stroke.50 An anti-edema effect of hypothermia at 35‚ĄÉ for 8-10 days has also been demonstrated in patients with intracerebral hemorrhage.51,52 ICP reduction does not necessarily equal improved clinical outcome. Nevertheless, therapeutic hypothermia is effective for ICP management, at least in selected patients (Figure 3). Currently, two major clinical trials of therapeutic hypothermia in patients with stroke are underway: The Intravenous Cooling in the Treatment of Stroke 2/3 (ICTuS 2/3) is a phase II-III trial of therapeutic hypothermia at 33‚ĄÉ for 24 hours in 1,600 stroke patients, and EuroHYP-1 is a phase III trial of therapeutic hypothermia at 34-35‚ĄÉ for 24 hours in 1,500 stroke patients.53 Despite the current lack of evidence from clinical trials on the neuroprotective effect of therapeutic hypothermia in stroke patients, the effect of therapeutic hypothermia on ICP reduction is unequivocal in patients with brain edema irrespective of the cause.54
In practice, therapeutic hypothermia can be separated into three phases: induction, maintenance, and rewarming. Induction is typically performed as rapidly as possible to a target temperature of 32-35‚ĄÉ. For this purpose, ice packs and 4‚ĄÉ cold saline can be combined with a commercialized cooling device. During the maintenance period, only minor fluctuations in patients' body temperature are allowed. The rewarming period is very important because rebound edema and ICP elevation may occur during this period.48 To avoid such detrimental effects, rewarming rates should be slow and are usually targeted at 0.1-0.25‚ĄÉ/h. Rebound hyperthermia after rewarming should also be avoided. Further clinical studies are required to determine the optimal therapeutic time window, target temperature, duration of cooling, and rewarming rate of therapeutic hypothermia for patients with stroke.
The prevention and management of various complications of therapeutic hypothermia is challenging. The most frequently encountered and cumbersome complication is shivering, especially when surface cooling is used rather than endovascular cooling.55 Shivering not only counteracts the cooling process but also has a harmful impact on systemic oxygen consumption, brain tissue oxygenation, and ICP.56,57 Our anti-shivering protocol developed according to a literature review and our clinical experience is shown in Table 4.58 The masking of infection-induced fever is also a problem associated with therapeutic hypothermia. We have experienced some patients whose circulating water temperature in the cooling device could be used as a surrogate marker for increased heat production by the patient. Regular follow-ups of chest radiographs and laboratory tests including complete blood cell counts, C-reactive protein, procalcitonin, urinalysis, and surveillance cultures may also help early detection of infection. Other major complications of therapeutic hypothermia include pneumonia, pulmonary edema, hypotension, decreased cardiac output, bradycardia, conduction blocks, ileus, hepatic dysfunction, pancreatitis, hyperglycemia, cold diuresis, hypokalemia, hypomagnesemia, hypophosphatemia, hypocalcemia, alkalosis, hypocarbia, impaired immune function, bleeding diathesis, and alterations in pharmacokinetics and pharmacodynamics. Physicians utilizing therapeutic hypothermia should be familiar with the prevention and management of such complications.
Ventilatory support
Most stroke patients with mild respiratory difficulties can be managed with supportive care such as insertion of an oral airway, supplemental oxygen, and antibiotics for aspiration pneumonia. However, patients who are stuporous or comatose following massive stroke can develop reduced oropharyngeal muscle tone that leads to posterior displacement of the tongue and airway obstruction. A decrease in or absence of gag and cough reflexes and impaired immune function further increase the risk of aspiration. Therefore, these patients are vulnerable to hypoxia, aspiration pneumonia, and respiratory distress and arrest unless preemptive endotracheal intubation and mechanical ventilation are applied. For safe intubation, RSI is necessary. RSI involves prompt induction of sedation and subsequent endotracheal intubation. Etomidate is the preferred induction agent for stroke patients because of its short-acting characteristics and hemodynamic stability. However, it is not recommended for patients with seizures or adrenal insufficiency. Other induction agents include propofol and midazolam. Succinylcholine is the most commonly administered neuromuscular blocking agent for RSI as a result of its rapid onset and short duration of action. However, a brief increase in ICP has been reported following administration of succinylcholine.59 Other possible side effects of succinylcholine are malignant hyperthermia, hyperkalemia, and aggravation of neuropathy or myopathy.59 For this reason, some experts recommend non-depolarizing neuromuscular blocking agents such as cisatracurium and rocuronium for RSI.59 Fentanyl can reduce the increase in blood pressure and ICP that occurs during intubation and is usually administered to stroke patients undergoing RSI, whereas lidocaine minimizes coughing and the increase in ICP.60
Intubated patients usually require mechanical ventilation. Inappropriate management of mechanically ventilated patients with space-occupying stroke can lead to lung damage as well as brain damage. Injuries to the lungs are attributable to a high end-inspiratory and low end-expiratory lung volume which results in repeated collapse and re-expansion. A high shear force is exerted on alveolar walls and small airways during inflation, especially at the interfaces between collapsed and aerated alveoli.61 Application of positive end-expiratory pressure (PEEP) can avoid repeated alveoli collapse. However, the effect of PEEP on cerebral physiology is controversial. PEEP can elevate ICP through increased thoracic pressure and decreased MAP, venous return, and cardiac output. One study increased PEEP up to 12 cmH2O in patients with acute stroke, but ICP remained unchanged or slightly reduced.62 There was marked decrease in CPP caused by the decrease in MAP. In general, increasing PEEP up to 20 cmH2O does not have deleterious effects on ICP as long as the baseline ICP is not high (<20 mmHg).63 The benefits of PEEP may outweigh the risks of hypoxemia in patients with stroke. A strategy of maintaining a low tidal volume and limiting plateau pressure is also necessary to restrict end-inspiratory overexpansion of alveoli. Tidal volume should be limited to 6-8 mL/kg of predicted body weight, especially for patients with acute respiratory distress syndrome (ARDS).64 However, low tidal volume reduces carbon dioxide elimination via the lungs and can cause hypercapnia and respiratory acidosis. Subsequent hyperventilation stimulates the brainstem respiratory center and may cause ventilator dyssynchrony and require heavy sedation or neuromuscular blockade. Hypercapnia causes intracranial hypertension by dilating cerebral vessels and increasing CBF. However, the risk of increased ICP as a consequence of permissive hypercapnia has not been studied in stroke patients.65 Pumpless extracorporeal lung assist combined with lung-protective ventilation may be used as an adjunctive therapy in patients with both ICP crisis and hypercapnia caused by low tidal volume.66 This can be used to address hypercapnia-associated increases in ICP, and the minute volume of the ventilator can be reduced to avoid hyperinflation of alveoli.
Sedation and analgesia
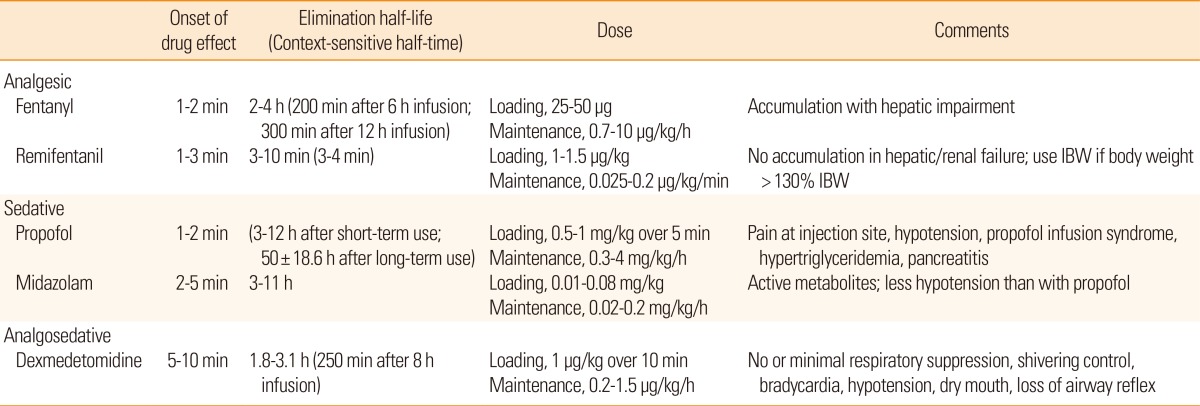
Sedation and analgesia are often required in patients with malignant brain edema following stroke. They can be used to manage ICP and CPP as well as attenuate the stress response, increase endotracheal tube tolerance, reduce metabolic energy demands, prevent delirium and decrease patient-ventilator synchrony.67 In the neurological ICU, sedation and analgesia are typically induced by continuous infusion of fentanyl, remifentanil, propofol, midazolam, or dexmedetomidine. Clinical studies that compare the efficacy of these agents are lacking in patients with stroke. In general, the different intravenous opioids are equally effective when titrated to similar pain intensity endpoints.68 Fentanyl, a ¬Ķ-receptor agonist, is popular in the ICU as a result of its strong analgesic effect, lack of neurotoxicity, rapid onset of action, and short elimination half-life. Remifentanil, an opioid agent that is chemically related to fentanyl, may be preferred to fentanyl as a result of its shorter elimination half-life and its lack of accumulation in patients with hepatic or renal failure. A short context-sensitive half-time-the time required for blood or plasma concentrations of a drug to decrease by 50% after discontinuation of drug administration-is an attractive characteristic of remifentanil when frequent wake-up tests for neurological evaluation and ventilator weaning are planned. Propofol has sedative, hypnotic, anxiolytic, amnestic, antiemetic, and anticonvulsant properties, but no analgesic effects.69 The rapid onset and short duration of action of propofol are advantageous if frequent wake-up tests are required and this agent may have stronger suppressive effects on cerebral metabolism than midazolam.70 However, propofol may lead to dose-dependent respiratory depression and hypotension. When administered at a high dose for a prolonged period (>4 mg/kg/h for >48 h), propofol may cause propofol infusion syndrome, which is characterized by various early signs including lactic acidosis, triglyceridemia, bradycardia, and Brugada-like electrocardiogram changes, and later signs including cardiac failure, tachyarrhythmia or heart block, ventricular tachycardia, rhabdomyolysis, hyperkalemia, renal failure, and fatty degeneration of the liver.71 Among the benzodiazepines, midazolam is often used for continuous infusion in the ICU as it has a faster onset, more rapid clearance, and shorter duration of action than lorazepam. However, with prolonged use, it exhibits greater variability and longer time to awakening than lorazepam. It also contains propylene glycol, which can cause metabolic acidosis and acute kidney injury.72 Dexmedetomidine is a selective alpha 2-receptor agonist with sedative, analgesic, and sympatholytic properties, but no anticonvulsive properties.73 Patients sedated with this agent are more easily aroused and more interactive than patients sedated with other drugs.74 Another characteristic of dexmedetomidine that is useful in the neurological ICU is the minimal respiratory depression and anti-shivering effects. In mechanically ventilated adult ICU patients at risk of developing delirium, dexmedetomidine infusion for sedation was associated with a lower prevalence of delirium than benzodiazepine infusion.75 However, dexmedetomidine frequently causes bradycardia, hypotension, and hypertension, especially after the loading dose. For this reason, we prefer starting with a maintenance dose of 0.5 ¬Ķg/kg/h without a loading dose. Dexmedetomidine is becoming increasingly popular in the neurological ICU. Physicians who manage stroke patients in the ICU should be familiar with the characteristics of each sedative and analgesic agent (Table 5).68
Neurological examination remains the most valuable tool for the assessment of stroke patients. The requirement for a reliable neurological examination conflicts with the requirement for sedation and analgesia in patients with massive stroke and mechanical ventilation. The sedation and analgesia regimen should utilize the minimum amount of drugs possible to adequately maintain a safe environment for ventilation and for cerebral and systemic hemodynamics during periods requiring tracking of neurological status.76 A regular wake-up test is an important evaluation measure in the neurological ICU. To perform this test, infusion of sedatives and analgesic agents should be interrupted temporarily. Precise assessment and neurological tracking can be helpful for planning neuroimaging studies and medical or surgical interventions. However, a wake-up test may cause an ICP surge, agitation, systemic desaturation, a decrease in brain tissue oxygen tension, and cerebral metabolic distress.76 Thus, neurological wake-up tests are controversial as a result of the risk of a stress response and the uncertainty of its clinical value. Early studies showed that a daily wake-up test was associated with a shorter ICU stay and a more positive outcome in general ICU patients.77,78 However, in more recent studies, evidence to support the overall benefit of a wake-up test has been equivocal.79,80,81,82 It is considered safe for medical and surgical ICU patients but has not been validated for patients in neurological ICU.77,78,83 In a prospective observational study, patients with acute brain injury developed a critical increase in ICP and impending brain tissue hypoxia as well as a sympathetic stress response with increased heart rate, respiratory rate, and blood pressure during interruption of sedatives for neurological examination.76 In a subgroup of patients with lower cerebral compliance, a wake-up test induced marked changes in ICP and CPP that could potentially have a negative impact on the injured brain. Thus, these patients need to be excluded from repeated wake-up tests, and information should instead be gathered from multimodal neuromonitoring in combination with neuroimaging studies.84 During a wake-up test, ICP and CPP often increase slightly and tolerably from baseline levels. In the majority of patients, these changes are mild, transient and acceptable, and do not preclude further repetitions of the test. Nevertheless, abrupt cessation of sedatives and analgesic agents is stressful to patients. A recent observational study showed that wake-up tests induced a biochemical stress response in patients with traumatic brain injury.85 Stress hormones including adrenocorticotrophic hormone, epinephrine, norepinephrine, and cortisol as well as physiological variables like blood pressure, heart rate, ICP, and CPP increased significantly immediately after wake-up tests.85 The short- and long-term consequences of the test are unclear at present. Therefore, it is important to consider the advantages and disadvantages of interrupting sedation and analgesia and stimulating patients for a wake-up test during an ICP crisis. Given the limited knowledge on the benefits of interrupting sedation and analgesia for wake-up tests, multimodal neuromonitoring and neuroimaging in neurocritical care may be helpful in unconscious patients with stroke.9
Once the clinical condition of the patient improves, sedatives and analgesics should be tapered and suspended. Definitive interruption is possible when the clinical and cerebral state of the patient do not justify sedation. However, acute withdrawal symptoms are not uncommon following abrupt cessation of sedatives and analgesics. This phenomenon is similar to paroxysmal sympathetic hyperactivity, i.e., episodic manifestations of fevers, diaphoresis, hypertension, tachycardia, tachypnea, and dystonic posturing.86 In this situation, sedatives and analgesics should be reinstituted and tapered slowly again after such symptoms disappear.
Conclusions
Brain swelling following stroke can cause secondary cerebral ischemia, herniation syndrome, and brain death. Aggressive medical and surgical treatment for brain swelling is necessary to prevent detrimental outcomes. General medical care is paramount because a complex interaction exists between the brain and other organs. The importance of total body care cannot be overemphasized: attention should be paid not only to the brain, but also to the other regions of the body. The advantages and limitations of each treatment should always be considered because the appropriate treatment for one area of the body may contradict the appropriate treatment for another.