Introduction
Acute cerebral ischemia elicits an immune response that leads to a cascade of events culminating in neuronal death and injury to supportive structures in the brain [1]. Several mechanisms, including excitatory toxicity, oxidative stress, inflammation, and apoptosis, are thought to be involved in the pathogenesis of ischemic stroke [2]. Additionally, neural stem cells (NSCs), which might contribute to the regeneration of the injured brain, can be damaged by ischemic injury [3]. Substantial clinical trials with drugs that modulate these mechanisms have failed to show efficacy after ischemic stroke.
GV1001 is a vaccine peptide anticancer agent derived from a 16-amino-acid sequence in the active site of human telomerase reverse transcriptase (hTERT). hTERT has both telomeric and extra-telomeric functions [4,5]. By mimicking the extra-telomeric function of hTERT, GV1001 has various biological functions, including antioxidant, anti-inflammatory, and anti-apoptotic effects, induction of cell proliferation, and mitochondrial stabilization. These effects can protect against ischemia-reperfusion injury (IRI) [3,6].
Based on the diverse effects of GV1001, we hypothesized that it might have neuroprotective effects against injury due to cerebral ischemia and reperfusion. In this study, we investigated the protective effect of GV1001 against ischemic stroke in an animal model and oxygen-glucose deprivation/reoxygenation (OGD/R)-derived injury in NSCs.
Methods
In vivo methods
Animal preparation and experimental protocols
All animal procedures were performed in accordance with the Hanyang University Guidelines for the Care and Use of Laboratory Animals and were approved by the Institutional Animal Care and Use Committee of Hanyang University. Male Sprague-Dawley rats weighing 280 to 300 g were used (Biogenomics Incorporated, Seoul, Korea). In accordance with the Animal Research: Reporting of In Vivo Experiments (ARRIVE) guidelines, all experiments were conducted in a strictly blinded fashion using predetermined inclusion and exclusion criteria and attrition due to mortality and other causes reported. However, sample sizes a priori could not be calculated because anticipated differences in infarct size and estimated efficacy with GV1001 were not available.
Middle cerebral artery occlusion of rats
After a 7-day period of adaptation and pre-training for neurological examination, the left middle cerebral artery was occluded for 2 hours and reperfused using the intraluminal filament technique described previously [7,8] and in the Supplementary methods.
Dose response and therapeutic time window of efficacy
In the first experiment to determine the most effective dose, rats were randomly divided into five groups: sham, saline, and different doses of GV1001 (6, 30, and 60 μM/kg)-treated groups (n=10 in each group). The sham group was subjected to the same protocol without middle cerebral artery occlusion (MCAO). Either saline or different doses of GV1001 (6, 30, and 60 μM/kg) were subcutaneously injected immediately before reperfusion. All rats were sacrificed 48 hours after MCAO, and the infarct volume was calculated after staining with 2,3,5-triphenyltetrazolium chloride (Sigma-Aldrich, St. Louis, MO, USA) as described previously [7]. Then, their brains were used for molecular biological assessment.
Magnetic resonance imaging-based cohort
After determining the optimal dose, another cohort randomized after diffusion-weighted imaging (DWI) was studied. The rats underwent DWI (repetition time [TR]/echo time [TE]=4,455/74 ms, slice thickness/gap=1/1 mm, matrix=64×63, and field of view [FOV]=50×50 mm) at 3-Tesla using an Achieva device (Philips, Best, The Netherlands) 2 hours after MCAO. DWI lesion volumes were measured using the Medical Image Processing, Analysis, and Visualization (MIPAV) software (National Institutes of Health, Bethesda, MD, USA). Rats were divided blindly to receive either saline (n=13), 30 μM/kg GV1001 (n=12), or 60 μM/kg GV1001 (n=12). Rats in the treatment and control groups were subjected to fluid-attenuated inversion recovery (FLAIR) magnetic resonance imaging (MRI) (TR/TE/inverstion time [TI]=11,000/77/2,800 ms, slice thickness/gap=1/1 mm, matrix=128×128, and FOV=50×50 mm) 48 hours after MCAO. The final infarct volume was independently assessed with MIPAV by neurologists blinded to the experimental groups and was calculated from the average of the two measurements (Figure 1A).
Transferase-mediated deoxyuridine triphosphate nick end labeling assay
To detect apoptotic cells caused by ischemic stroke, a transferase-mediated deoxyuridine triphosphate nick end labeling (TUNEL) assay (Roche Boehringer Mannheim, Indianapolis, IN, USA) was performed. The sham group underwent the same protocol without MCAO, and the saline group was treated with saline. GV1001 was administered at doses of 30 and 60 μM/kg. The detailed method for the TUNEL assay is described in the Supplementary methods.
Neurological outcomes
1) Beam walking test
The sensorimotor function was assessed using beam walking, rotarod, and modified sticky-tape tests performed before MCAO and 2 and 3 days after MCAO by an investigator blinded to the experimental groups. For the beam walking test, rats were trained 7 days before MCAO to walk on a wooden beam (2.5×2.5×80 cm) placed 60 cm above the floor to return to their home cage. The test was scored as follows: 0, traverses the beam with no foot slip; 1, traverses by grasping the lateral side of the beam; 2, shows disability walking on the beam but can traverse; 3, takes a considerable amount of time to traverse the beam because of difficulty in walking; 4, unable to traverse the beam; 5, unable to move the body or any limb on the beam; 6, unable to stay on the beam for 10 seconds [9].
2) Rotarod test
Rats were placed on an accelerating rotarod cylinder, and the riding time that the animal remained on the rotarod was measured. The speed of the rotarod was not increased during the experiment. A trial ended if the animal fell off the rungs or gripped the device and spun around for two consecutive revolutions without attempting to walk on the rungs. The animals were trained 7 days before MCAO. Motor test data are presented as the mean duration (three trials) on the rotarod [9,10].
3) Modified sticky-tape test
A sleeve was created using a 3 cm piece of green paper tape that was 1 cm wide. The tape was wrapped around the forepaw so that the tape attached to itself and allowed the fingers to protrude slightly from the sleeve that was formed. The typical response was for the rat to vigorously attempt to remove the sleeve by either pulling at the tape with its mouth and/or brushing the tape with its contralateral paw. After attaching the sleeve, the rat was placed in its cage and observed for 30 seconds. The timer was turned on only when the animal attempted to remove the taped sleeve. The data collected represented the fraction of the 30 seconds observation period that the animal spent attending to the stimulus. The contralateral and ipsilateral limbs were tested separately. The test was repeated three times per test day, and the best two scores were averaged [11].
Histology and immunohistochemistry
All rats used for the evaluation of infarction volume with MRI were sacrificed 48 hours after MCAO for histological and immunohistochemical analyses. Hematoxylin and eosin (H&E) staining distinguished the peri-infarct region from the remaining area. TUNEL-positive cells were counted in the GV1001 and control (sham and saline) groups. Immunohistochemical staining was performed as previously described [7-9] using antibodies against phosphorylated Akt (pAkt; Ser473, 1:100, 9271, Cell Signaling Technology, Beverly, MA, USA), phospho-glycogen synthase kinase (pGSK-3β; 1:100, ab107166, Abcam, Cambridge, MA, USA), phosphorylated-extracellular signal-regulated kinase (pERK)1/2 (Thr202/Tyr204; 1:1,000, 9101, Cell Signaling Technology), B-cell lymphoma 2 (Bcl-2; 1:100, sc-7382, Santa Cruz Biotechnology, Dallas, TX, USA), Bcl-2 associated X (Bax; 1:100, ab5714, Abcam), nestin (1:200, ab6142, Abcam), neuronal nuclei (NeuN; 1:100, MAB377, Millipore, Bedford, MA, USA), doublecortin (DCX; 1:100, ab28941, Abcam), and SRY-box transcription factor 2 (SOX2; 1:50, ab79351, Abcam; 1:50, ab97959, Abcam). The detailed methods are described in the Supplementary methods.
Western blotting
The peri-infarct region in the control (sham and saline) group and the corresponding area in the GV1001-treated group, which were confirmed based on MRI, were used in Western blot analysis for phospho-insulin receptor substrate-1 (pIRS-1) (Ser636/639; 1:1,000, 2388, Cell Signaling Technology), phospho-phosphoinositide 3-kinase (pPI3K) p85 (Tyr458)/p55(Tyr199) (p85α PI3K; 1:1,000, 4228, Cell Signaling Technology), pAkt (Ser473), Akt (1:2,000, 9272, Cell Signaling Technology), pGSK-3β (1:1,000, 9336, Cell Signaling Technology), GSK-3β (1:2,000, sc-9166, Santa Cruz Biotechnology), pERK1/2 (Thr202/Tyr204), Bcl-2, Bax (1:1,000, 2772, Cell Signaling Technology), uridine diphosphate glucuronosyltransferase 1A1 (UGT1A1, 1:500, ab237810, Abcam), FK506 binding protein 5 (FKBP5, 0.4 μg/mL, NBP1-84676, Novus Biologicals, Centennial, CO, USA), sphingosine kinase type 1 (SPHK1, 1:1,000, ab71700, Abcam), and beta-tubulin (β-tubulin, 1:2,000, 2146, Cell Signaling Technology). The detailed method for Western blotting is described in the Supplementary methods.
In vitro methods
Culture and treatment of NSCs and primary cortical neurons
NSCs were isolated from rodent embryonic brains, cultured, and then expanded. NSCs were cultured as described previously [12,13]. Primary cultured cortical neurons were obtained from the cerebral cortex of fetal Sprague-Dawley rats (Orient Bio, Seongnam, Korea) on embryonic day 16. The methods for culturing NSCs and primary cortical neurons are briefly described in the Supplementary methods.
OGD was achieved in a Forma anaerobic chamber (Thermo Fisher Scientific, Waltham, MA, USA). A gas mixture containing 5% CO2, 0.2% O2, and 94.8% N2 was flushed through the chamber for 1, 2, 4, 6, 8, 10, 12, 14, 16, 22, and 24 hours. This procedure maintained a non-fluctuating hypoxic environment of <1% O2. Reoxygenation was performed after 1 and 8 hours of incubation in a hypoxic chamber, and the cells were incubated again in a normoxic incubator (95% air/5% CO2) for 8 hours at 37°C. Untreated cells served as controls.
To evaluate the effect of GV1001 on NSCs, the cells were treated with several concentrations of GV1001 alone. Finally, to measure the protective effect of GV1001 against OGD, NSCs were treated with several concentrations of GV1001 under OGD for 1 and 8 hours, as described above.
Molecular studies
The diverse molecular analyses included (1) trypan blue staining (TBS) and lactate dehydrogenase (LDH) release assay to measure cell viability and cytotoxicity; (2) bromodeoxyuridine (BrdU) cell proliferation assay; (3) colony-forming unit (CFU) assay to determine cell growth; (4) annexin V and propidium iodide staining to detect cell death; (5) evaluation of apoptosis; (6) determination of cell migration; (7) determination of free radical production; (8) malondialdehyde (MDA) assay of lipid peroxidation due to oxidative stress; (9) determination of Ca2+ level; (10) determination of oxidative mitochondrial DNA damage; (11) adenosine triphosphate (ATP) assay; (12) mitochondrial membrane potential assay; (13) Western blot analysis; (14) proteomics analysis; and (15) antibody microarrays. The methods are detailed in the Supplementary methods.
Statistical analyses
All data are presented as the mean±standard deviation values from five or more independent experiments. Statistical analyses of three or more datasets were performed using one-way analysis of variance, followed by Tukey’s post hoc comparisons. Statistical significance was set at P<0.05. All statistical analyses were performed using IBM SPSS Statistics for Windows version 21.0 (IBM Co., Armonk, NY, USA).
Results
In vivo results
Reduction of infarct volume by GV1001
Administration of GV1001 2 hours after MCAO attenuated the infarct volume at 48 hours. The effective doses of GV1001 were 30 and 60 μM/kg, which were used in subsequent experiments (Figure 1B). The initial infarct volumes on DWI were not significantly different between the GV1001-treated and saline-treated groups (P=0.278). The infarct volume on FLAIR MRI 48 hours after MCAO compared to lesion volume on DWI MRI just before injection showed a significantly smaller ratio in the GV1001-treated group than in the saline-treated group (Figure 1C).
Efficacy of GV1001 based on behavioral evaluation and expression of TUNEL-positive cells
The GV1001-treated groups showed consistently better neurological outcomes than the saline group. The treatment effects over time were significant in a dose-dependent manner for the rotarod, modified adhesive-removal, and beam balance tests (Figure 1D). After sacrificing the rats, brain tissues were sectioned and analyzed by TUNEL assay to assess apoptotic cell death. The number of TUNEL-positive cells in the ischemic periinfarct region was compared among the sham, saline, and GV1001-treated groups (30 and 60 μM/kg GV1001). The number of TUNEL-positive cells was reduced in the GV1001-treated group compared with the control group in a dose-dependent manner (Figure 1E).
Effect of GV1001 on intracellular signals
To investigate the mechanisms underlying GV1001-induced neuroprotection against ischemic stroke, the immunoreactivity of pIRS-1, pAkt (Ser473), pGSK-3β (Ser9), pERK1/2, Bcl-2, Bax, glial fibrillary acidic protein (GFAP, an astrocyte marker), nestin (a neuroectodermal stem cell marker), NeuN (a neuronal nuclear antigen), DCX (a neuronal differentiation marker), and SOX2 (a multipotent NSC marker) were measured. Immunoreactivities of survival-related proteins (pIRS-1, pAkt, pGSK-3β, pERK1/2, and Bcl-2) were significantly increased in the periinfarct region after treatment with GV1001. The expression of Bax, which is involved in cell death, was decreased by treatment with GV1001 (Figure 2A).
Immunohistochemistry confirmed the increases in pAkt (Ser473), pGSK-3β (Ser9), pERK, and Bcl-2 levels and decreases in Bax levels along with increment in the number of NeuN- or SOX2-positive cells after treatment with GV1001 (Figure 2B and Supplementary Figure 1). Additionally, increased levels of nestin, NeuN, SOX2, and DCX were noted in the peri-infarct subventricular zone of the GV1001-treated group, but GFAP expression was decreased by GV1001 treatment (Figure 2C). The levels of markers for neurotoxic phenotype reactive astrocytes (UGT1A1 and FKBP5) were increased and those of neuroprotective phenotype reactive astrocytes (SPHK1) were decreased in the peri-infarct area of the MCAO rat model, and GV1001 significantly restored the expression of these markers (Figure 2D).
In vitro results
Effect of OGD/R and GV1001 on NSC viability, proliferation, and mobilization
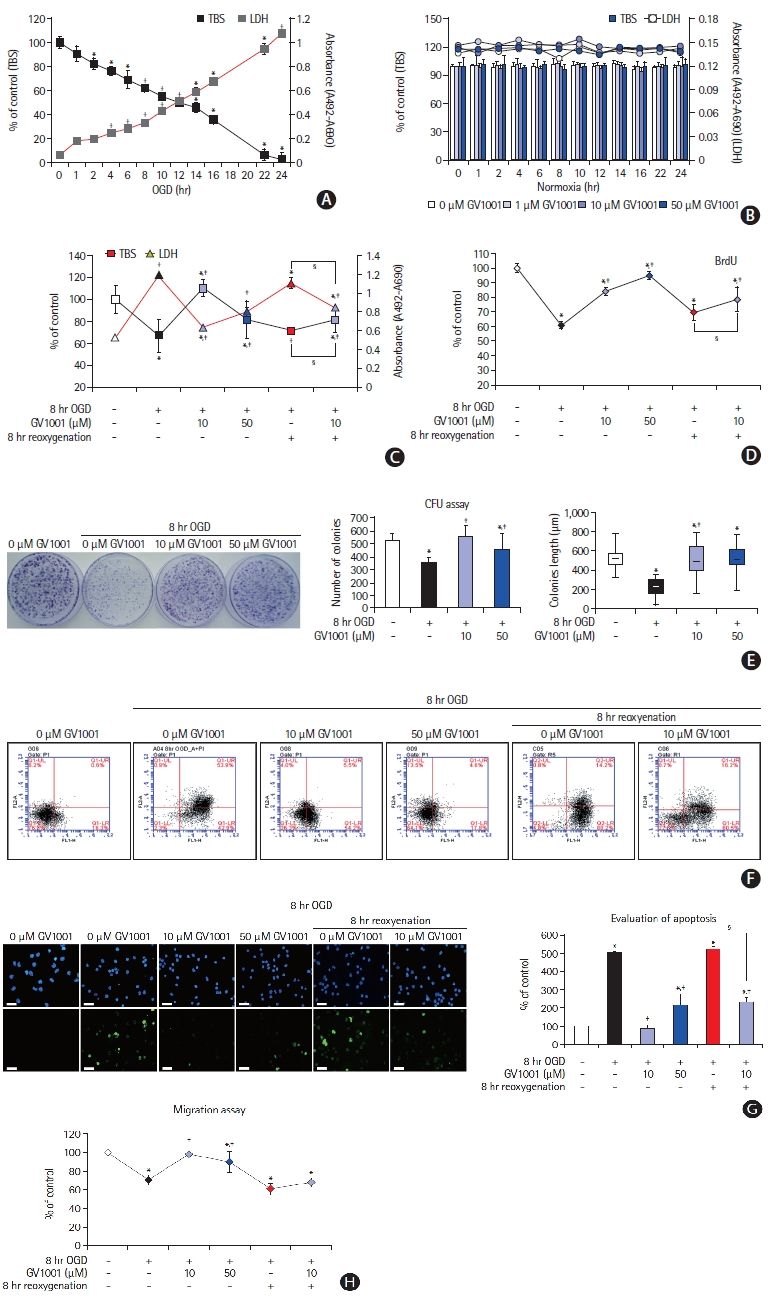
Cell viability was measured using TBS and LDH assays. Exposure of primary cultured NSCs to OGD for 1 to 24 hours significantly reduced NSC viability in a time-dependent manner (Figure 3A). As cell viability was 60% to 70% after 8 hours of OGD exposure, this condition was considered as optimal duration (Figure 3A). GV1001 alone did not affect the viability of NSCs (Figure 3B).
NSCs were exposed to OGD for 8 hours and treated simultaneously with different concentrations of GV1001. Compared with treatment with OGD/R alone, treatment with OGD/R and GV1001 at concentrations of 10 and 50 μM increased NSC viability, as measured by the TBS and LDH assays (Figure 3C and F), and proliferation, as measured by the BrdU and CFU assays (Figure 3D and E).
The effect of GV1001 on the apoptosis of NSCs induced by OGD/R was determined by 4',6-diamidino-2-phenylindole (DAPI) and TUNEL staining. These results revealed that the percentage of apoptotic cells was increased by OGD/R and was significantly decreased by co-treatment with GV1001 (up to 50 μM) (Figure 3G). Migratory capacity, which is an important characteristic of stem cells, was evaluated using a migration assay kit. OGD/R reduced the migratory capacity of NSCs, but co-treatment with GV1001 (10 and 50 μM) effectively restored this capacity (Figure 3H).
Effect of GV1001 on free radical production and oxidative mitochondrial DNA damaged by OGD/R
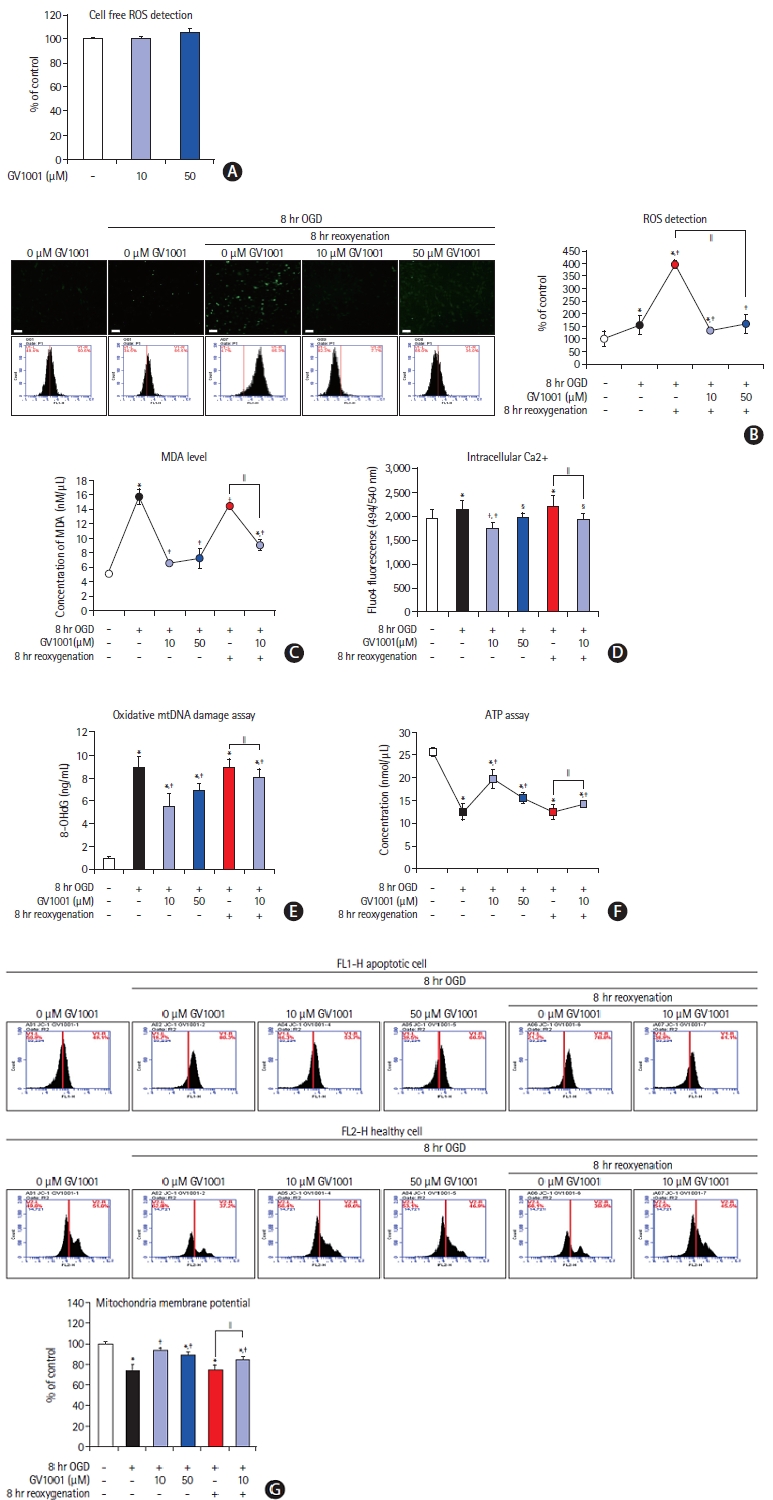
We evaluated the free radical scavenging effect of GV1001 after OGD. GV1001 alone did not trigger reactive oxygen species (ROS) production under cell-free conditions (Figure 4A). ROS levels were dramatically elevated by reoxygenation after OGD in NSCs, and GV1001 significantly inhibited ROS production (Figure 4B). The levels of MDA, a lipid peroxidation marker, were measured to confirm the antioxidant effect. The MDA levels in NSCs were increased by OGD/R. However, GV1001 effectively decreased this level in a concentration-dependent manner (Figure 4C). Intracellular Ca2+ levels decreased, and oxidative mitochondrial DNA damage increased with OGD/R. These effects were significantly reversed by the administration of GV1001 (Figure 4D and E).
Effect of GV1001 on mitochondria of NSCs injured by OGD/R
We measured ATP levels and mitochondrial membrane potentials to evaluate the effect of GV1001 on the mitochondria of NSCs injured by OGD/R. ATP levels were reduced in NSCs after OGD/R, but this reduction was significantly alleviated by GV1001 treatment (Figure 4F). The mitochondrial membrane potential in NSCs was decreased by OGD/R but was significantly restored by GV1001 (Figure 4G).
Effect of OGD/R and GV1001 on intracellular protein levels
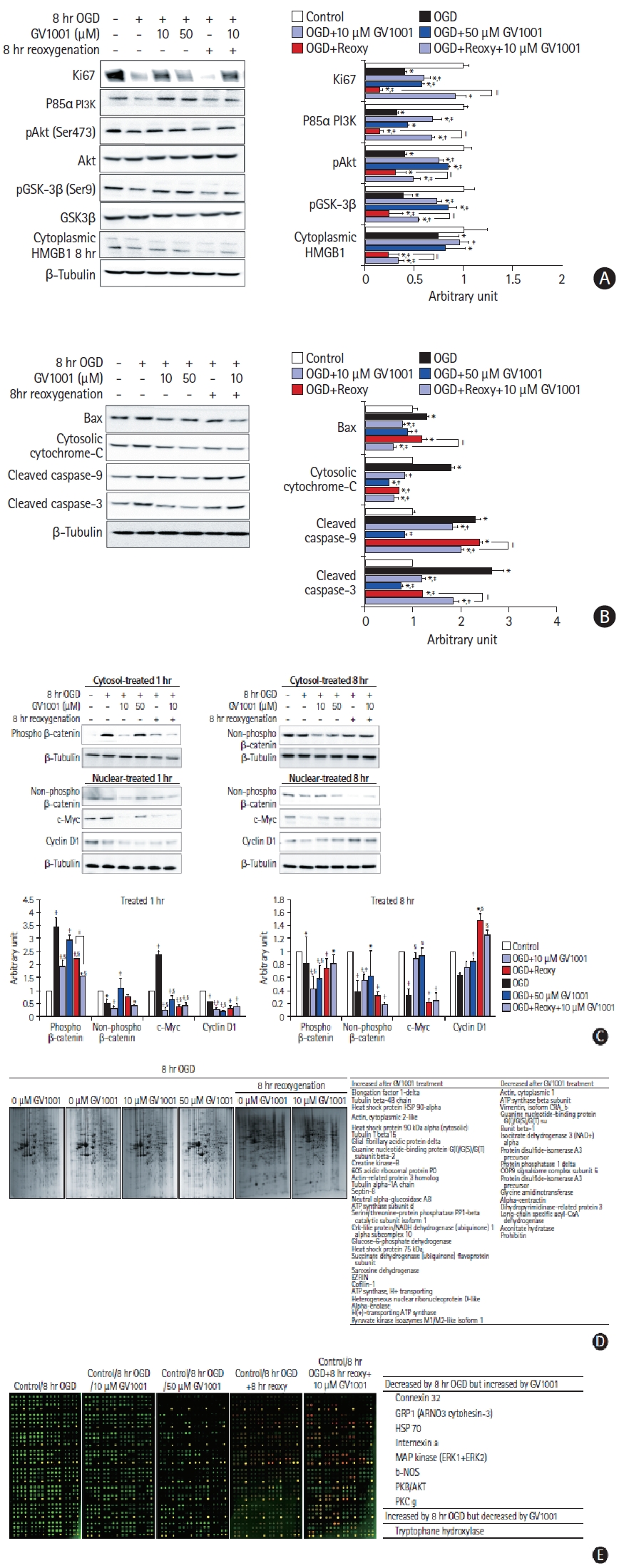
We performed Western blot and immunocytochemistry analyses to directly measure changes in the immunoreactivities of proteins affected by treatment with OGD/R and GV1001 (Figure 5A-C). Ki67 level, which is associated with proliferative activity, was decreased by OGD/R but was increased by GV1001 (Figure 5A). The levels of survival-related proteins, such as p85α, PI3K, pAkt (Ser 473), pGSK-3β (Ser9), and cytoplasmic high mobility group box protein 1 (HMGB-1), were also decreased following OGD/R injury but were recovered with GV1001 treatment (Figure 5A). The levels of death-related proteins, such as Bax, cytosolic cytochrome c, cleaved caspase-3, and cleaved caspase-9, were increased with OGD/R but were significantly decreased with GV1001 treatment (Figure 5B).
We also measured alterations in the Wnt signaling pathway to examine the non-canonical telomerase activity. OGD/R decreased active (non-phospho) β-catenin expression in the cytoplasm, whereas GV1001 significantly increased active β-catenin expression. OGD/R significantly decreased the expression of cyclin D1 and c-Myc, but GV1001 effectively restored their expression, although the expression pattern of c-Myc was restored slightly at 1 hour after treatment (Figure 5C). Additionally, proteomic analyses were performed to determine the effect of OGD/R and GV1001 on the production of intracellular proteins in NSCs. The production of numerous proteins was altered by OGD/R, and treatment with GV1001 restored the altered protein production (Figure 5D). An antibody microarray was performed to assess the phosphorylation patterns of proteins. The levels of the phosphorylated forms of various survival-related proteins were decreased by 8 hours of OGD/R and were restored by GV1001 treatment. The level of tryptophan hydroxylase, which catalyzes serotonin synthesis, was increased by OGD/R and decreased by GV1001 treatment (Figure 5E).
Critical roles of PI3K pathway in GV1001-induced rejuvenation of stem cell characteristics of OGD/R-injured NSCs
OGD/R significantly deteriorated the stem cell characteristics, including survival, proliferation, and migration of NSCs. However, GV1001 effectively counteracted these effects and restored the PI3K pathway. To investigate the role of the PI3K pathway in the rejuvenation of NSCs by GV1001, NSCs were pretreated with the PI3K inhibitor LY294002 before OGD/R and GV1001 treatments. Pretreatment with the PI3K inhibitor blocked the effect of GV1001 on OGD-injured NSCs, and cell viability was significantly decreased compared to that of NSCs not pretreated with the PI3K inhibitor (Figure 6). However, in NSCs injured with OGD/R (8 hours OGD with 8 hours reoxygenation), the PI3K inhibitor attenuated the protective effect of GV1001, but the effect was not statistically significant.
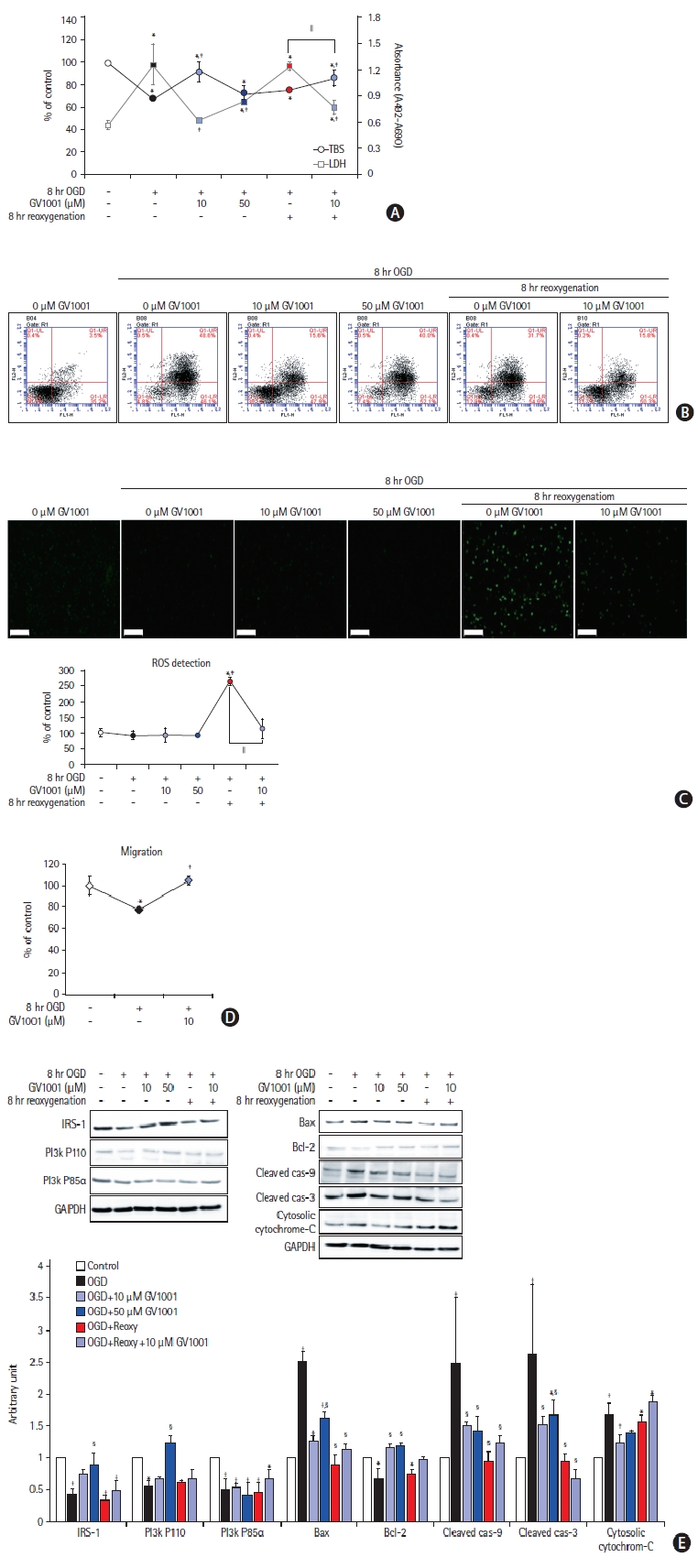
Effect of GV1001 on cortical neurons damaged by OGD/R
To confirm the effect of GV1001 on neurons injured by OGD/R, viability, toxicity, and ROS production were assessed (Figure 7). GV1001 restored the viability and reduced the toxicity of cortical neurons, which were injured by OGD/R (Figure 7A and B). Furthermore, GV1001 significantly inhibited the production of ROS, which was elevated by reoxygenation after OGD, in these neurons (Figure 7C). The levels of survival-related proteins were decreased and those of death-associated proteins were increased by OGD/R. However, treatment with GV1001 reversed these changes (Figure 7D).
Discussion
In the present study, we demonstrated that GV1001 reduced the infarct volume and restored the neurologic function in an ischemic stroke rat model. GV1001 stimulated neuroprotective pathways, as demonstrated by the increased levels of pAkt, pGSK-3β, pERK1/2, and Bcl-2, and decreased levels of Bax along with an increase in the number of NeuN-positive neurons and SOX2-positive NSCs. The expression of death-related proteins, including cleaved caspase-3, cleaved caspase-9, and Bax, was suppressed by GV1001. The in vitro OGD/R model demonstrated that GV1001 protected NSCs from OGD/R injury by improving viability, restoring proliferation, increasing mobilization, stabilizing mitochondria, and exerting antioxidant effects. In addition, GV1001 improved viability, increased migration activity, and decreased ROS in OGD/R-injured cortical neurons.
GV1001 is a vaccine peptide derived from hTERT [14], which was originally developed as an anticancer agent [15]. Recent studies have shown extra-telomeric functions of hTERT, including stabilizing mitochondria, promoting cellular proliferation, and antioxidant, anti-inflammatory, and anti-apoptotic effects [4,16]. GV1001 may protect against IRI by mimicking these extra-telomeric functions. In line with our finding that GV1001 showed protective effects against IRI in the brain, previous reports have described its protective effects against IRI in other organs. GV1001 significantly increased the survivability of skin flaps against IRI by reducing ROS levels and inflammation in a rat model [16]. Furthermore, GV1001 showed potential protective effects in a myocardial IRI rat model [17], lung transplantation rat model [18], and renal IRI mouse model [14]. However, to our knowledge, the present study is the first to show significant neuroprotective effects of GV1001 against cerebral IRI.
The PI3K pathway is important for cell survival and contributes to the protection of the brain and NSCs after ischemic injury [19]. Activated PI3K phosphorylates Akt downstream [20], and pAkt inactivates glycogen GSK-3β by phosphorylation and inhibits caspase-9, which is related to apoptosis [21,22]. Activated GSK-3β is associated with cell death due to ischemic injury [8]. In the present study, the in vitro OGD/R model confirmed the neuroprotective mechanism of GV1001, which was achieved by increasing levels of the survival-related proteins (PI3K, pAkt, and pGSK-3β) and decreasing levels of apoptosis-related proteins such as Bax, cytosolic cytochrome c, cleaved caspase-3, and cleaved caspase-9 (Figure 5A and B). These protective effects of GV1001 were significantly reduced by co-treatment with the PI3K inhibitor, LY294003 (Figure 6). In cortical neurons, GV1001 also increased the levels of survival-related proteins (IRS-1 and PI3K) and attenuated those of apoptosis-related proteins. We previously demonstrated the downstream PI3K pathway in cortical neurons after GV1001 treatment [5]. These findings indicate the direct effect of GV1001 on the PI3K pathway. Similar to the results of the in vitro OGD/R model, the PI3K pathway was associated with the neuroprotective mechanisms of GV1001 in an ischemic stroke rat model. These neuroprotective mechanisms were achieved by increasing pAkt, pGSK-3β, pERK1/2, and Bcl-2 levels and by suppressing Bax expression (Figure 2A and B), all of which are related to the PI3K pathway [21,23]. Proteomics revealed that GV1001 increased the levels of diverse intracellular proteins, including elongation factor 1-delta, heat shock protein 90α, and ezrin in OGD/R-injured NCSs. These proteins can promote the PI3K pathway [24-26]. Additionally, microarray data showed an increase in Akt expression with GV1001 treatment. After activation, NSCs differentiate into neural cells that replace damaged cells during neurogenesis [27]. The replacement of cells by regeneration of endogenous NSCs might restore neurological function after ischemic stroke [27,28]. However, endogenous NSCs are injured by ischemic stroke and lose their critical stem cell characteristics. SOX2 is a marker for multipotent NSCs [29]. In the peri-infarct subventricular zone of the stroke rat model, SOX2 levels were markedly increased in the GV1001-treated groups compared with the saline group (Figure 2C). An in vitro OGD/R model revealed that treatment with GV1001 restored the viability (Figure 3C) and proliferation (Figure 3D and E) activity of OGD/R-injured NSCs. GV1001 showed an anti-apoptotic effect (Figure 3G) and recovered the impaired migratory activity (Figure 3H). In addition, GV1001 demonstrated an antioxidant effect by reducing ROS levels (Figure 4B) and decreasing oxidative mitochondrial damage caused by OGD/R (Figure 4E). MDA and intracellular Ca2+ levels were assessed to verify the antioxidant effect of GV1001. GV1001 attenuated both MDA and intracellular Ca2+ levels, which were increased by OGD/R (Figure 4C and D).
The expressions of active β-catenin, cyclin D1, and c-myc were analyzed to evaluate the Wnt signaling pathway. This pathway has neuroprotective effects against and regenerative effects after ischemic injury [30]. In this study, active β-catenin, cyclin D1, and c-myc levels were decreased in NSCs following OGD/R injury but were restored by treatment with GV1001 (Figure 5C). These findings suggest that GV1001 has non-canonical telomerase activity, consistent with that seen in our previous study [5]. Additionally, levels of Ki67, which reflects proliferative activity [31], and those of HMGB-1, which modulates DNA repair and chromatin modification after DNA damage [32], were reduced by OGD/R, and treatment with GV1001 increased the levels of both (Figure 5A). Therefore, GV1001 may repair the DNA.
NSCs, which are activated under various conditions, differentiate into diverse types of nerve cells, including neurons, astrocytes, and oligodendrocytes [27]. After a stroke, NSCs more frequently differentiate to form astrocytes, which contribute to gliosis, with less frequent differentiation to neurons [27]. In this study, incremental increases in the levels of neuronal markers and decreases in the levels of astrocyte markers were evident in the peri-infarcted regions of the brains of GV1001-treated rates (Figure 2B). These findings can be explained by our previous report that GV1001 restores the ability of injured NSCs to differentiate more frequently to neurons and less frequently to astrocytes [5].
This study had two limitations. First, the pharmacokinetics of GV1001 were not evaluated. Second, no single animal model can reflect the heterogeneous nature of human stroke. Therefore, it is difficult to generalize the findings of the current study directly to ischemic stroke in humans.
Conclusions
In conclusion, we demonstrated the neuroprotective effect of GV1001 in an acute ischemic stroke animal model, as evidenced by improved tissue and neurological outcomes. GV1001 displayed neuroprotective effects against OGD/R injury in NSCs by inducing proliferation, restoring cell migration, reducing apoptosis, and stabilizing the mitochondria. These effects mimic the extra-telomeric functions of hTERT. Thus, GV1001 is a potential therapeutic candidate for ischemic stroke; however, further studies are needed to confirm this.